GROMACS Wizard – Step 1: Prepare

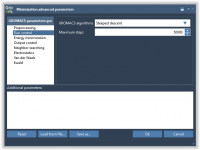
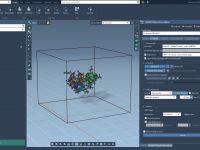
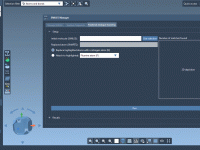
This section is part of the GROMACS Wizard tutorial. Once the system has been preprocessed and validated, we can start the preparation step. To launch GROMACS simulations, we first need to prepare the system: Choose the model (force field and solvent). Choose the molecular system and, optionally, choose a set of conformations or a path for a batch project. Define the periodic box. Choose what and how many ions should be added. Run preparation During the preparation step, GROMACS Wizard…