





This section is part of the GROMACS Wizard tutorial.
Once a system or a batch project has been successfully prepared, it is necessary to perform the Energy Minimization (EM) step to ensure that the system has no steric clashes or inappropriate geometry.
Switch to the Minimize tab.
Selecting input structure
When launching the Energy Minimization step, GROMACS Wizard requires you to provide one of the following:
- The path to a GRO file resulting from the previous step: either a GRO file resulting from the Preparation step or the previous launch of the Energy Minimization step.
- The path to a batch project, that has been prepared or resulted from one of the other steps.
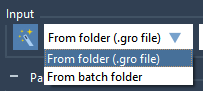
If you want to proceed from the previous step you can simply click on the Auto-fill button highlighted in the image below. This will automatically fill the path based on the previous project, whether a GRO file or a batch project from the previous successful run (e.g. from the Preparation step).
You can also choose the input GRO file yourself by clicking on the “…” button.
Choosing parameters
GROMACS molecular dynamics parameters for energy minimization are present in the Parameters section of the Minimize tab. By default, these parameters are populated with default values that are suitable for usual energy minimization runs. You can modify these parameters as needed. In the Parameters section, you will find the parameters that are most likely to be changed often, like the energy minimization tolerance.
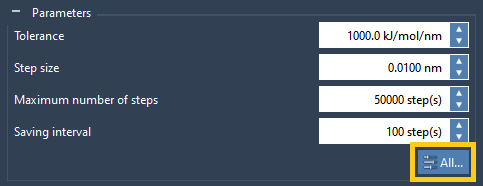
The other GROMACS molecular dynamics parameters can be accessed by clicking on the “All…” button highlighted in the image above. To learn more on how to apply custom parameters please check the Applying custom parameters section.
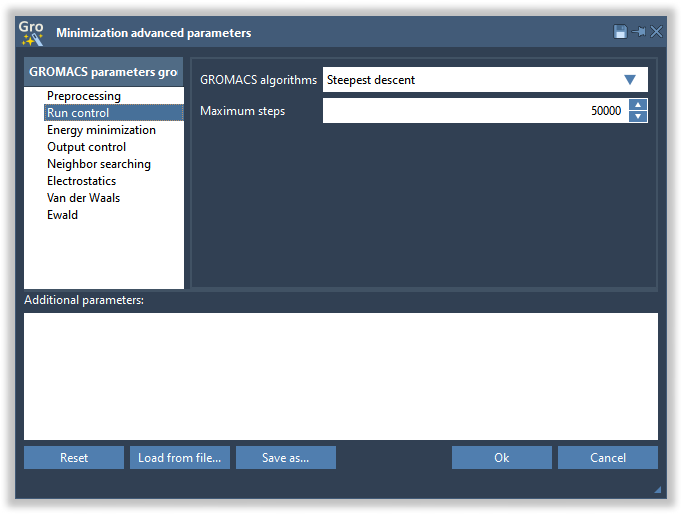
For the sake of this tutorial, leave the parameters to their default values.
To restore the parameters to their default values click on the “Reset” button in the Advanced parameters window. To load the parameters from an MDP file from some other project click the “Load from file…” button. To save the parameters in an MDP file click the “Save as…” button.
Note: the modified parameters are saved on closing SAMSON, so the next time you will have the previously saved parameters.
Run energy minimization
GROMACS Wizard gives you several possibilities:
- Generate inputs – generates a ready-to-use project that you can, for example, run on your local cluster.
- Minimize locally – launches the computations locally on your PC using the shipped GROMACS or the one you specified in the settings (see Using custom GROMACS version and performance parameters).
- Minimize in the cloud – launches the computations in the cloud. This is suitable for minimizing big systems that would otherwise require too many resources to minimize locally. This will prompt you to select the Cloud machine, etc. Note that launching in the Cloud requires computing credits. See Launching Computations in the Cloud for more information.
Now, simply click on the Minimize locally button to launch the Energy Minimization calculations on your PC. Some pop-ups might appear informing you about the current steps or possible warnings/issues if there are any.
Depending on your PC, the energy minimization of the system in the tutorial might take from several seconds to about one minute. You can see the current progress in the Output window that should pop up.
During these calculations, you can still use SAMSON and GROMACS Wizard thanks to the job manager of the GROMACS Wizard Extension. You can always access the list of the local GROMACS jobs and their state via the “Local jobs” button.
Importing the results
If you launched a single project and there are no other projects in the local job queue then after the computations are done a pop-up will appear asking for import options. If you launched multiple jobs or a batch job then you can check their state in the Local jobs. You can choose whether to import the whole trajectory, only the last frame, or some range of frames, what type of Periodic Boundary Condition treatment to apply, and on what to center the system. For example, as shown in the image below, you can choose to import only the last frame and to center the system on the Protein.
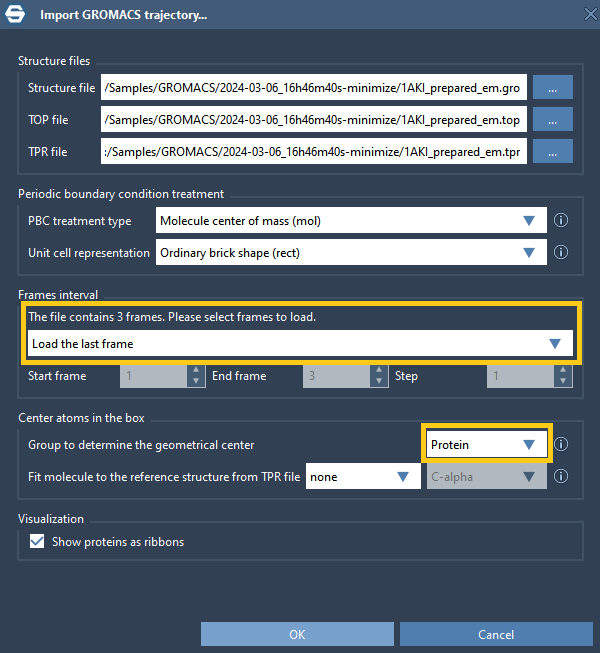
If you do not want to import the trajectory you can simply click Cancel – this will not delete any results and will still generate the plot.
You can access the results in the results folder specified at the top of the GROMACS Wizard. The folders with results are named with the launch date and time, and the step description (for the Energy Minimization step, the folder suffix is “_em“).
Plots
You can check the plot describing the evolution of the system’s potential energy, Epot, over the Energy Minimization steps at the bottom of the tab in the Plots section.
In this example, this plot demonstrates the nice, steady convergence of the potential energy.
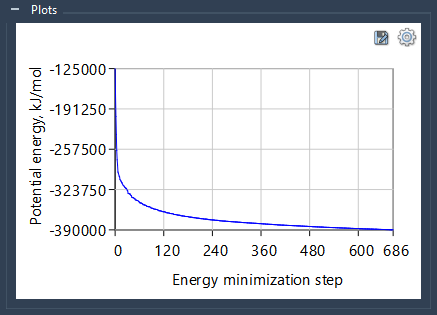
The plots are automatically generated and saved when the job is finished and the results are loaded. This plot is also generated for each subproject in a batch automatically. If you would like to save the plot, click on the Save button on top of the figure.
Checking results
There are two important factors to check to determine if Energy Minimization was successful and has converged:
– Potential energy – Epot, which should be negative, and (for a simple protein in water) on the order of 105-106, depending on the system size and number of water molecules.
– Maximum force – Fmax, the target for which is set in the advanced parameters of the Energy Minimization step (see the description below). For example, “emtol = 1000.0” indicates a target Fmax of no greater than 1000 kJ mol-1 nm-1. Please see the tooltips in the advanced parameters.
Please check the output for the potential energy and maximum force – they should be written per step and at the very end of the output. You can check them in the Output window.
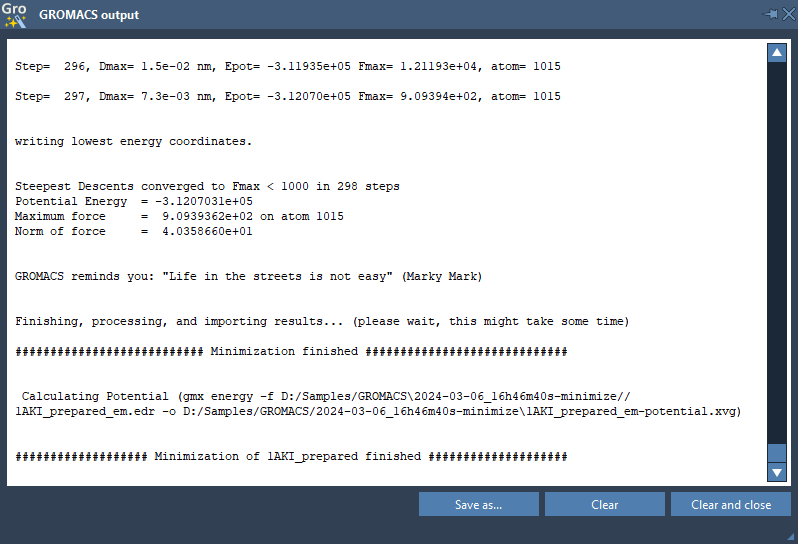
It is possible that Energy Minimization would reach the given maximum number of the Energy Minimization steps and will arrive at a reasonable Epot but with Fmax > emtol. If this happens, your system may not be stable enough for further equilibration and simulation. You might need to evaluate why it may be happening and, for example, increase the number of minimization steps (or run minimization again by providing the input data from the previous minimization step), or change the minimization parameters (integrator, emstep, etc).
| Previous: GROMACS Wizard – Step 1: Prepare | Next: GROMACS Wizard – Step 3: NVT Equilibration |