





In this tutorial, we will show you how to use the Protein Aligner SAMSON extension.
First, go to SAMSON Extensions web page, log in, and add the Protein Aligner SAMSON extension.
This Protein Aligner extension can align both sequences and structures of proteins.
Open SAMSON and use the Protein Data Bank Downloader to fetch two proteins: 1DLW and 1RTX. Those two are hemoglobin proteins from different organisms.
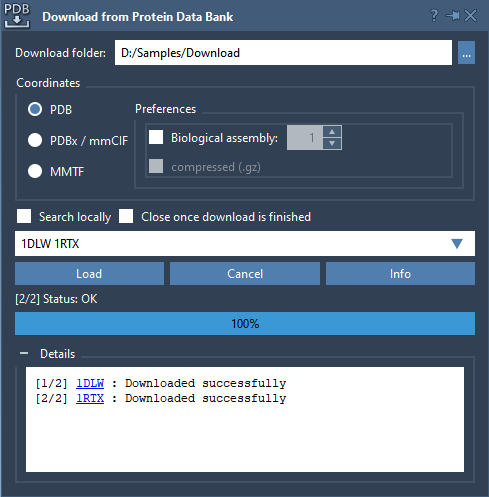
Now, click twice on the protein aligner editor ![]() to open the GUI.
to open the GUI.
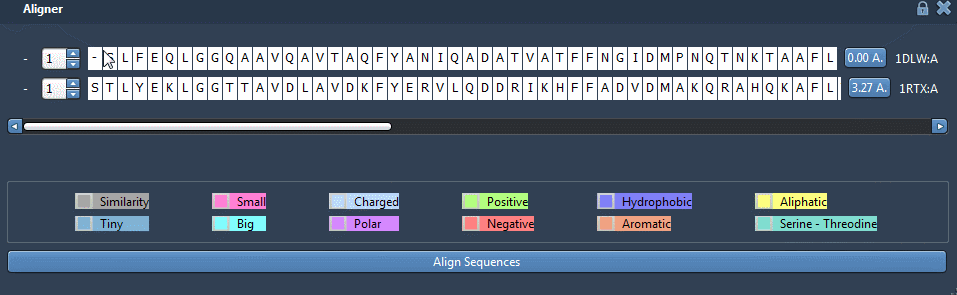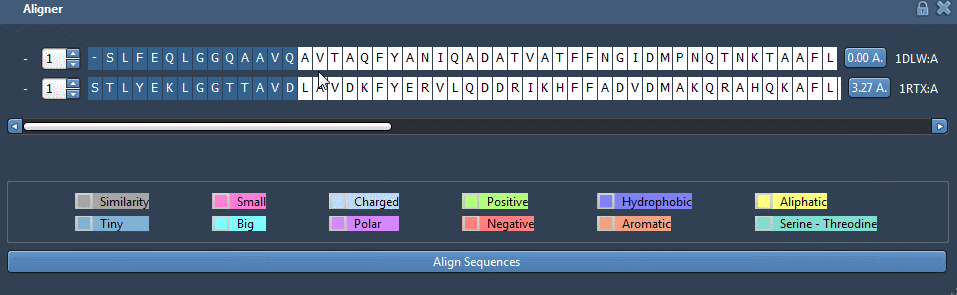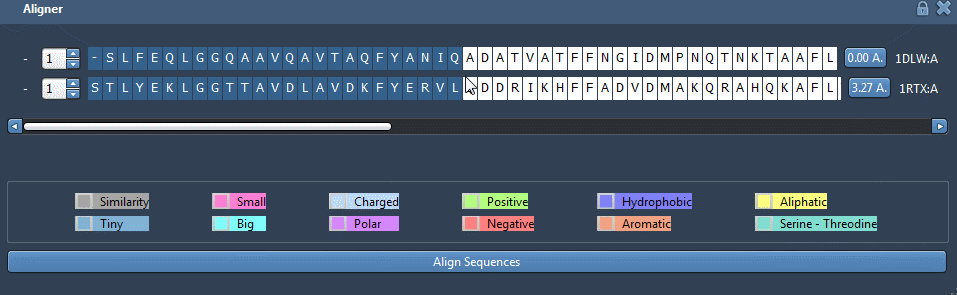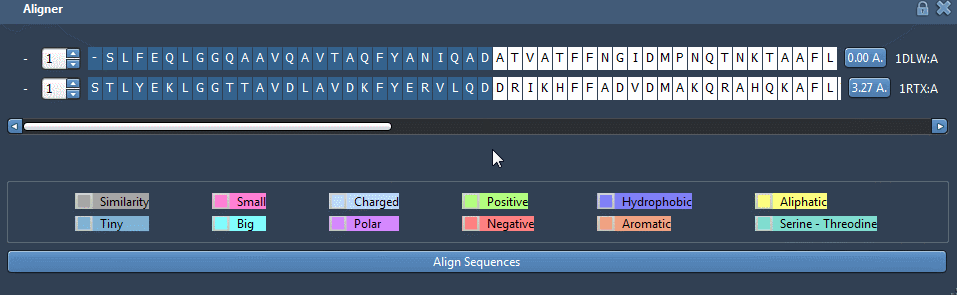
Click on “Align Sequences” and you should see the sequence alignment has been made like this:
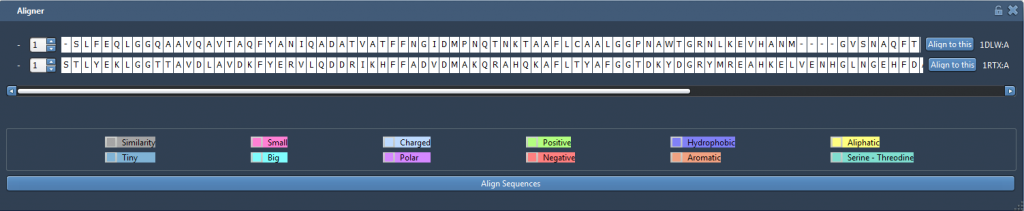
You can lock the window by clicking on the padlock.
Check some properties checkbox to see if the corresponding amino acids have the same properties. Here, we see that aromatic and aliphatic residues are quite well conserved between the two proteins:
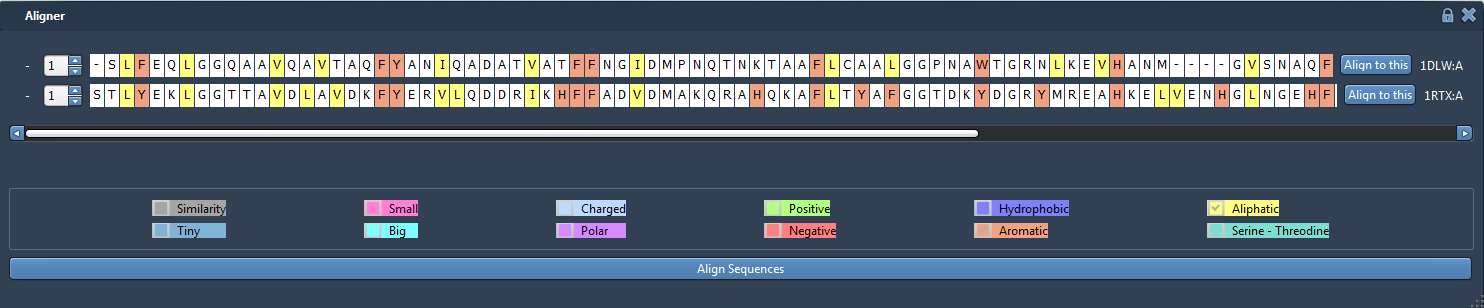
The residues in the aligner bar are selectable and are linked to the residues in the viewport. For example, you can try to select some residues marked as Hydrophobic and check that those residues are buried in the protein.
You can also visualize directly the conserved residues by clicking on “Similarity”.
Now, let’s perform a structural alignment of those two proteins. Make sure that no residue is selected then click on “Align to this”, on the first row.
Now, the other button indicates the distance between the two proteins: 3.27A. The two structures should be superimposed like this:
For now, we cannot see much from the atomistic representation. We can select one of the two proteins to differentiate them but it’s still not very clear. Let’s display the secondary structures of the proteins to have a better view:
- Select the two proteins
- Click on Visualization>Add Visual Model (or Ctrl+Shift+V)
- Choose Secondary structure and click on Ok
Now, hide the atomistic representation by unchecking the boxes in front of the proteins.
It should now look like that:
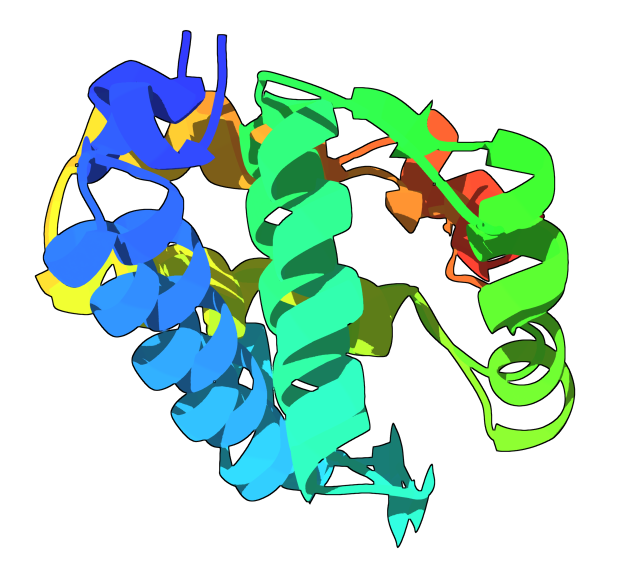
You can see from this picture that the two blue alpha-helices look very similar but are not well aligned. To align those two parts in particular, go back to the GUI and select the twenty first residues like this:
Click on the button 0.0A to superimpose specifically those residues of the structure.
That’s all for the tutorial of this module, now, just a few tricks that can be useful:
- While the editor window is active, selecting a residue in the document view will automatically select all the aligned residues in other structures.
- If you don’t want to deal with the aligner window, just select the aligner editor, select a protein, right-click on it. The contextual menu gives an option to align this protein on other present structures, and the RMSD will be displayed on the status bar.
- When you select some sequence in the aligner window, you can copy-paste it to a test editor in FASTA format.
If you have any questions or feedback, please use the SAMSON forum.