





Introduction
We previously proposed ([1]) a new implementation of the UFF interaction model introduced by Rappe et al. in [2], combined with an automatic scheme to perceive the molecular system in order to compute the bonds, bond orders, and atoms’ types. If interested, you can read our previous post about this interaction model.
This post is about the Interactive Modeling Universal Force Field (IM-UFF) interaction model that was published in [3] and which extends the UFF force field for performing interactive modeling. This extension of UFF is able to smoothly handle topological changes of the modeled systems. More specifically, IM-UFF allows for the creation and breaking of covalent bonds, changes in the order of covalent bonds as well as changes of atom typizations. With this extension, molecular structures can go through significant modifications while being simulated or edited, in order to reach a topology that better reflects the current organization of the involved atoms. Such an extension, incorporated in an interactive modeling process, lets the user easily build and edit molecular systems while being guided by physically-based inter-atomic forces.
Requirements
- IM-UFF Extension
Setting IM-UFF
The setting of IM-UFF interaction model is very close to the setting of UFF.
- Open a document with a molecular system you want to simulate with IM-UFF.
- Add a simulator by choosing Simulation, Add simulator in the main menu (or use the shortcut Ctrl+Shift+M).
- Select “Interactive Modeling Universal Force Field” in the list of interaction models.
- Choose the state updater you want to rely on for the simulation.
- Push the “OK” button.
Contrarily to UFF, there is no setup window in IM-UFF. You should now see the parameter window of IM-UFF. The parameter window of IM-UFF is close to the one of UFF. The main difference is the “Interactive modeling options” group on the top of the window which proposes two options:
- “Static topology (UFF only)”. This option allows one to switch between standard UFF when the checkbox is checked and IM-UFF when it is not. Important note: there is one major difference between “standard UFF” as implemented here in the IM-UFF interaction model and UFF in the previous Universal Force Field interaction model. It is that the bond energy in “standard UFF” is equal to “zero” when the atoms are at the infinite distance, whereas in UFF they are equal to “zero” when they are at equilibrium distance. This shift of energy was added to have equal energies with UFF and with IM-UFF for a given equilibrium state and because “zero” energy reference in IM-UFF corresponds to atoms non-interacting together. For more details, see [3].
- “Keep vdW for manipulated”. This option is active only with IM-UFF (i.e. when “Static topology” is unchecked). When it is checked, all the vdW energy and forces are computed whereas when it is unchecked, the atoms manipulated with the mouse are not considered while computing vdW interactions. This last setting makes manipulations easier since typically when you want to connect the manipulated atom to other atoms and vdW repulsive forces may hinder these connections.
Running IM-UFF simulation
- Be sure that the “Static topology (UFF only)” and the “Keep vdW for manipulated” checkboxes are unchecked.
- Press the “Simulation running” (green arrow) button.
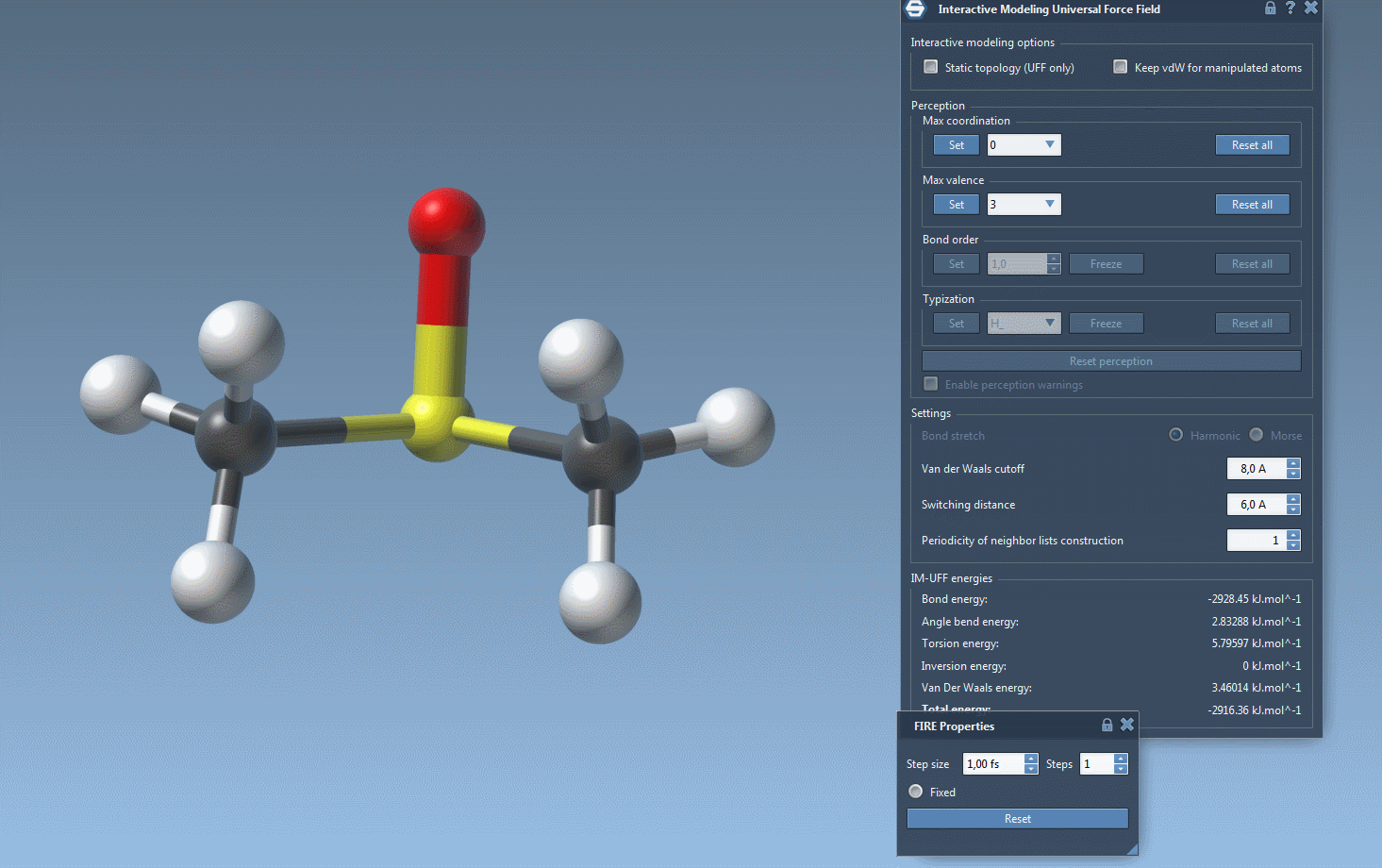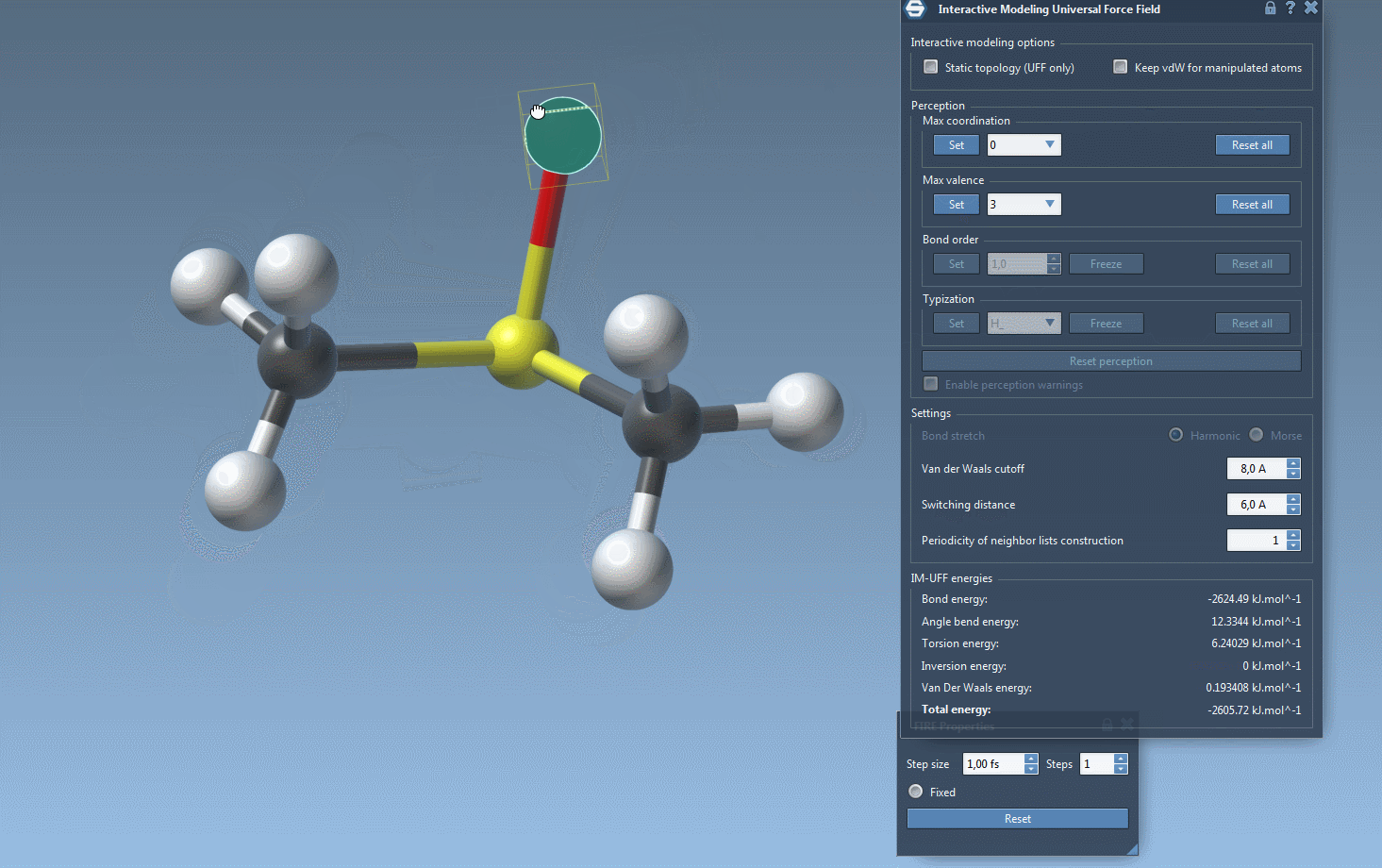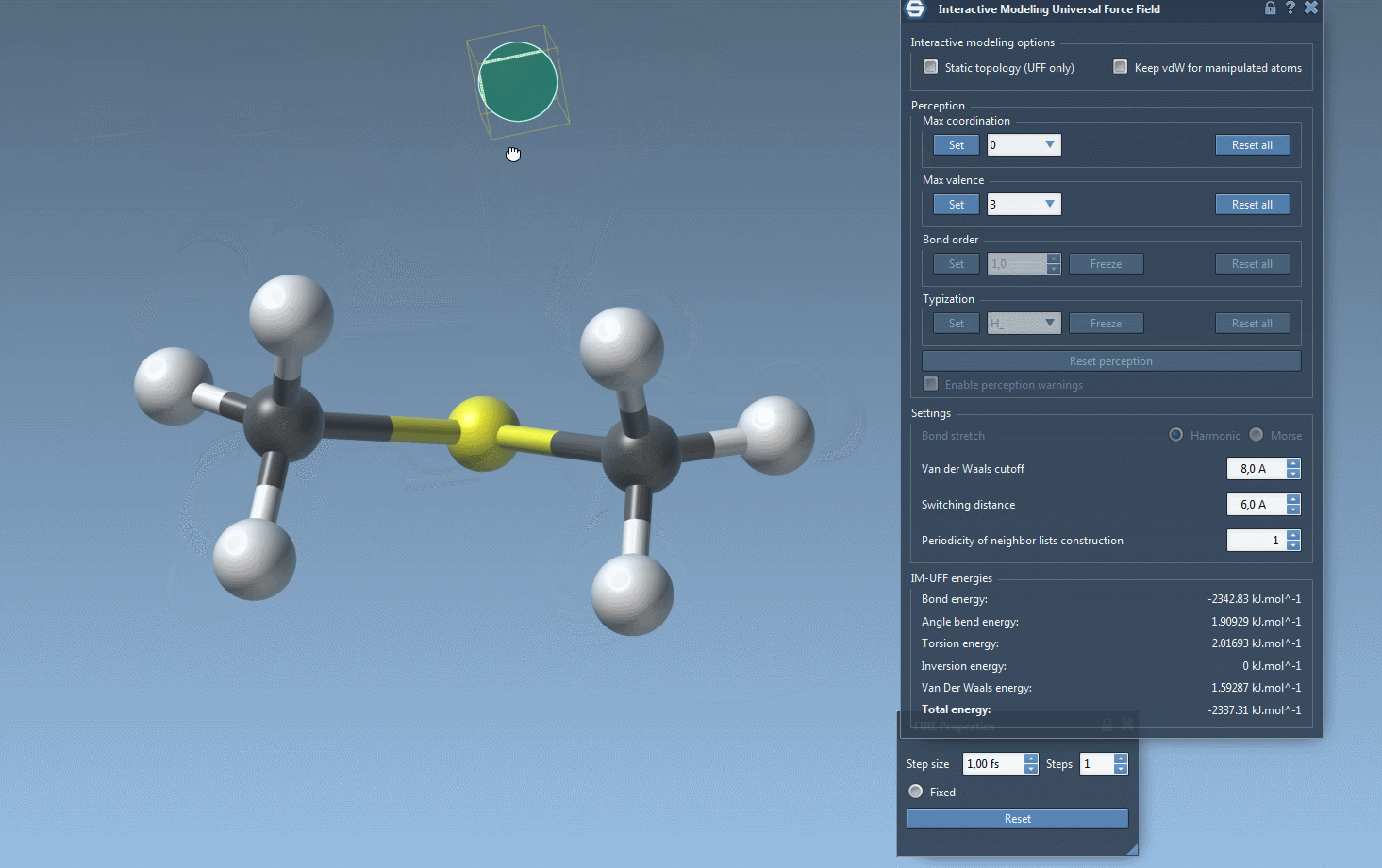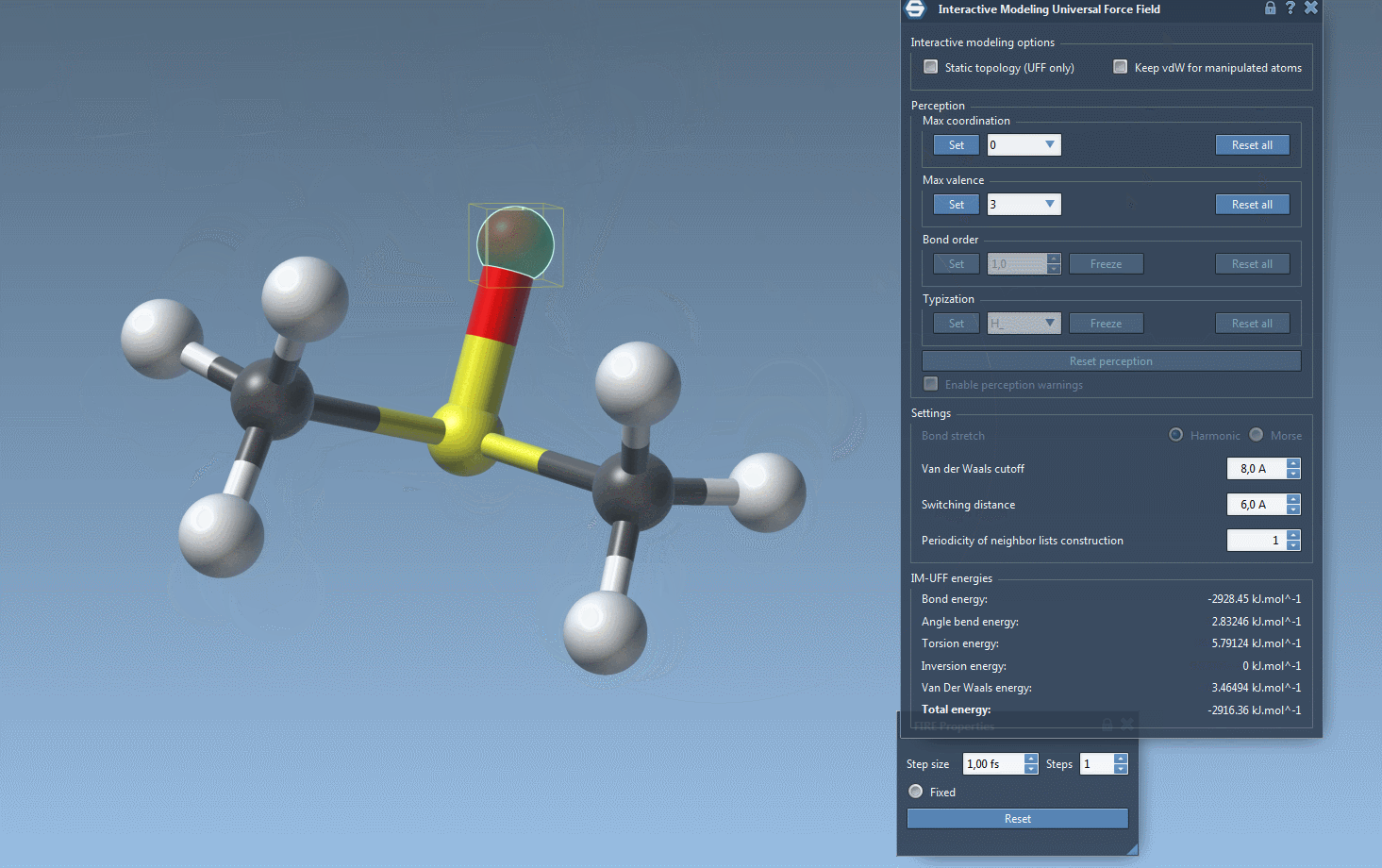
IM-UFF simulation is now running. You can see at the bottom of the IM-UFF parameter window the total energy of the system as well as each individual energy term. Try to displace the atoms with the mouse. You see that when you move slightly the atoms, the bonds are not broken and the system moves in order to locally adjust the structure while the topology is preserved. However, if you displace an atom a lot, it breaks the bonds to its neighbors and the topology is updated in consequence. On the contrary, when the atom location gets closer to some atoms, bonds can form to create and a new topology can appear.
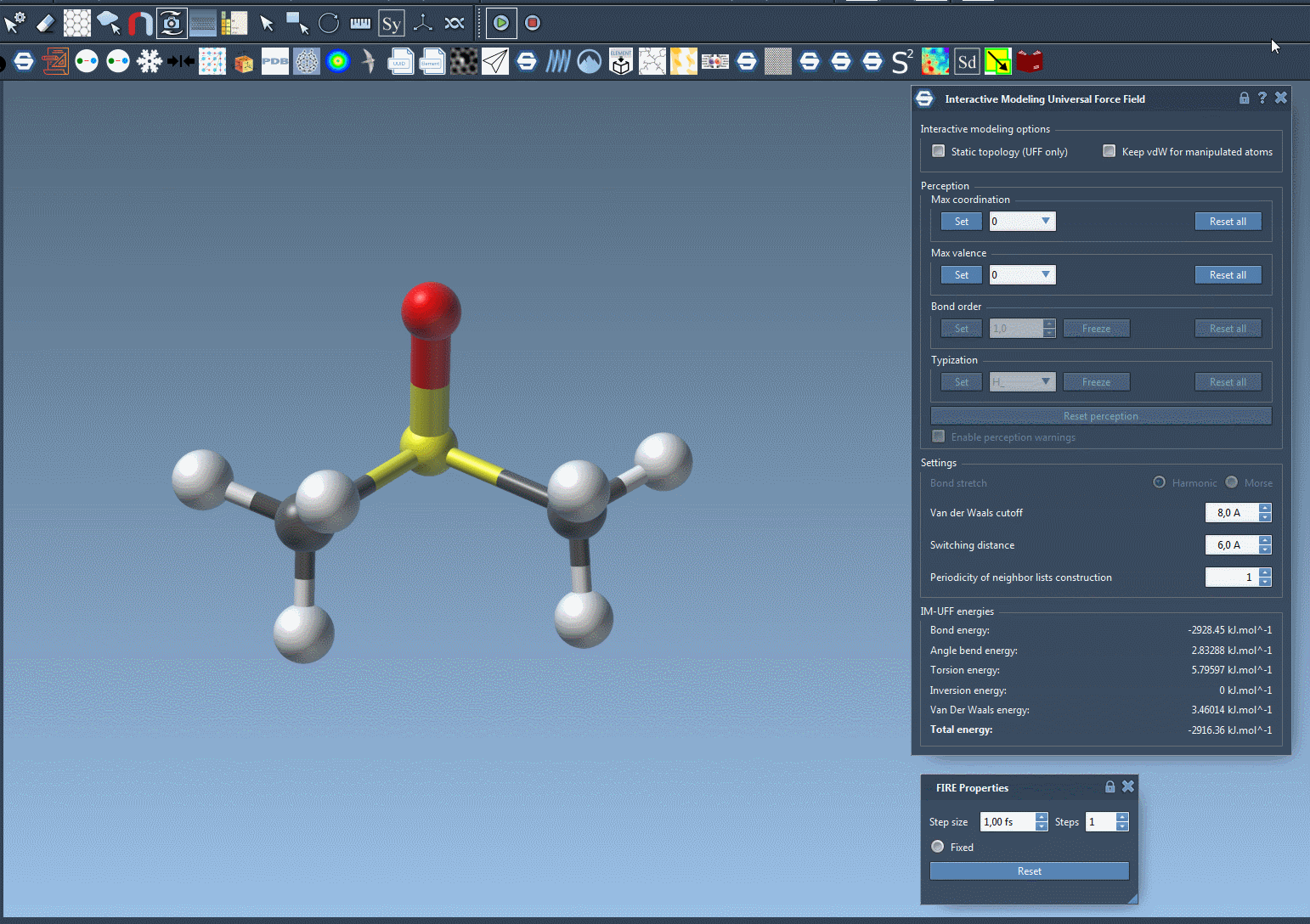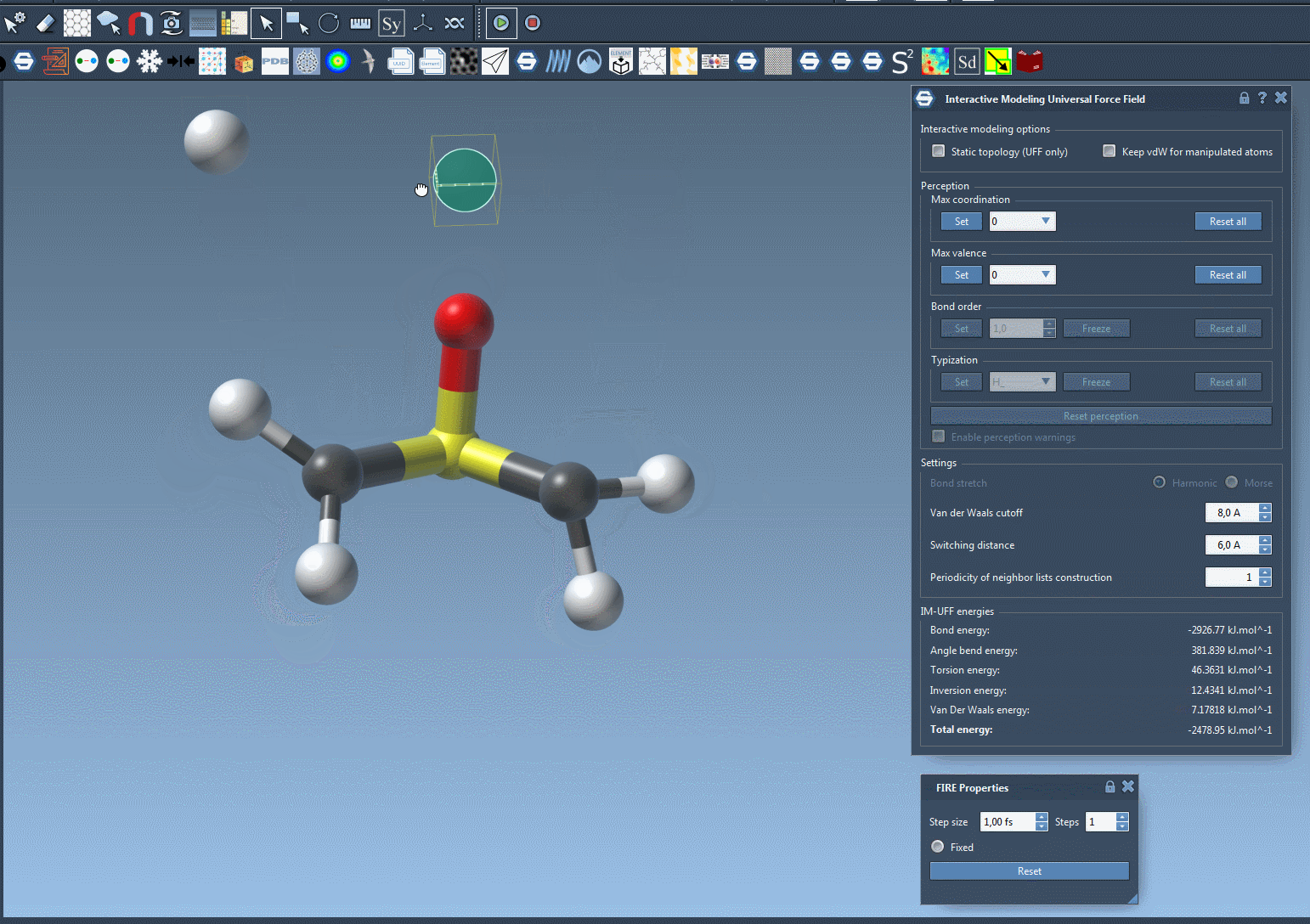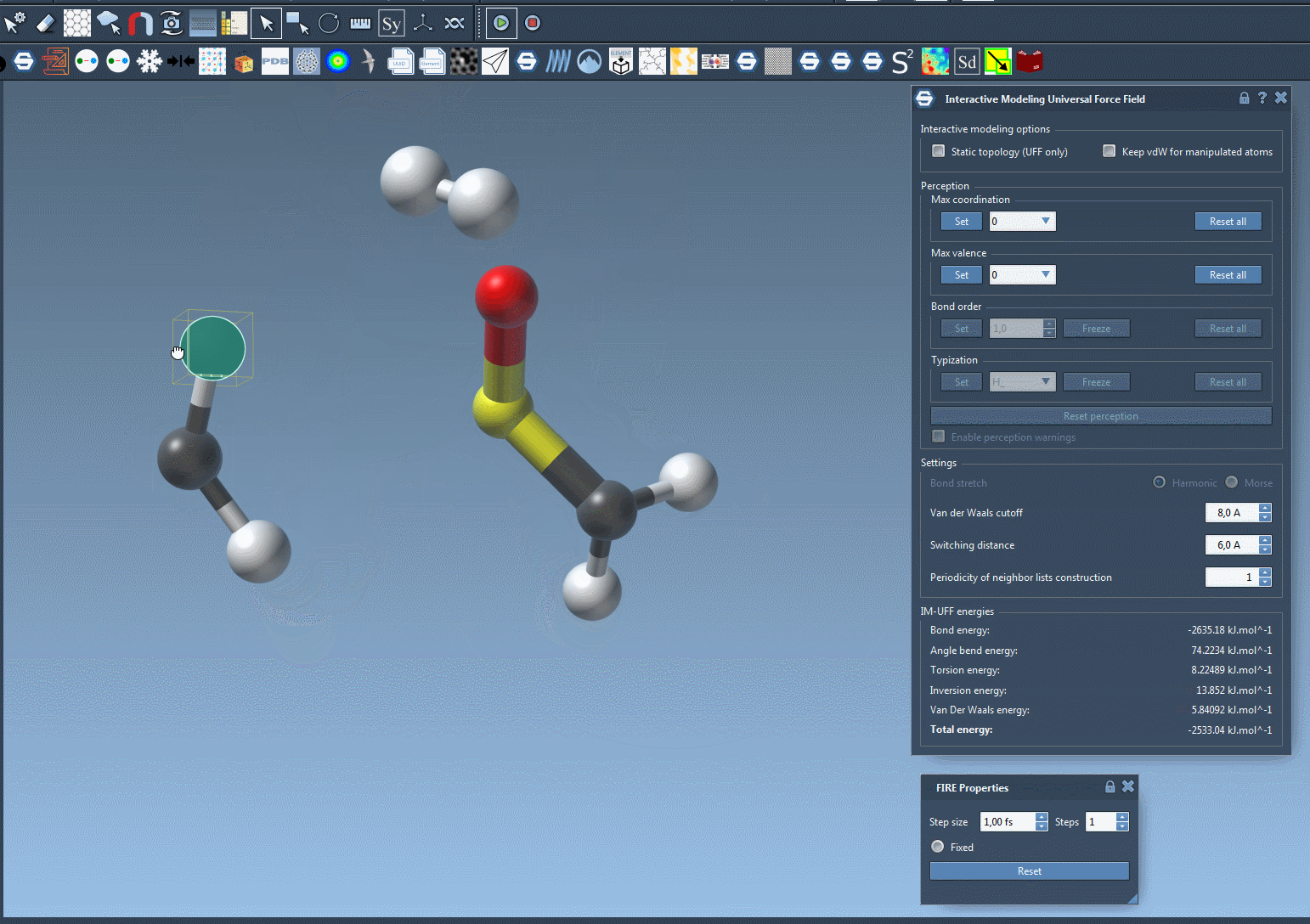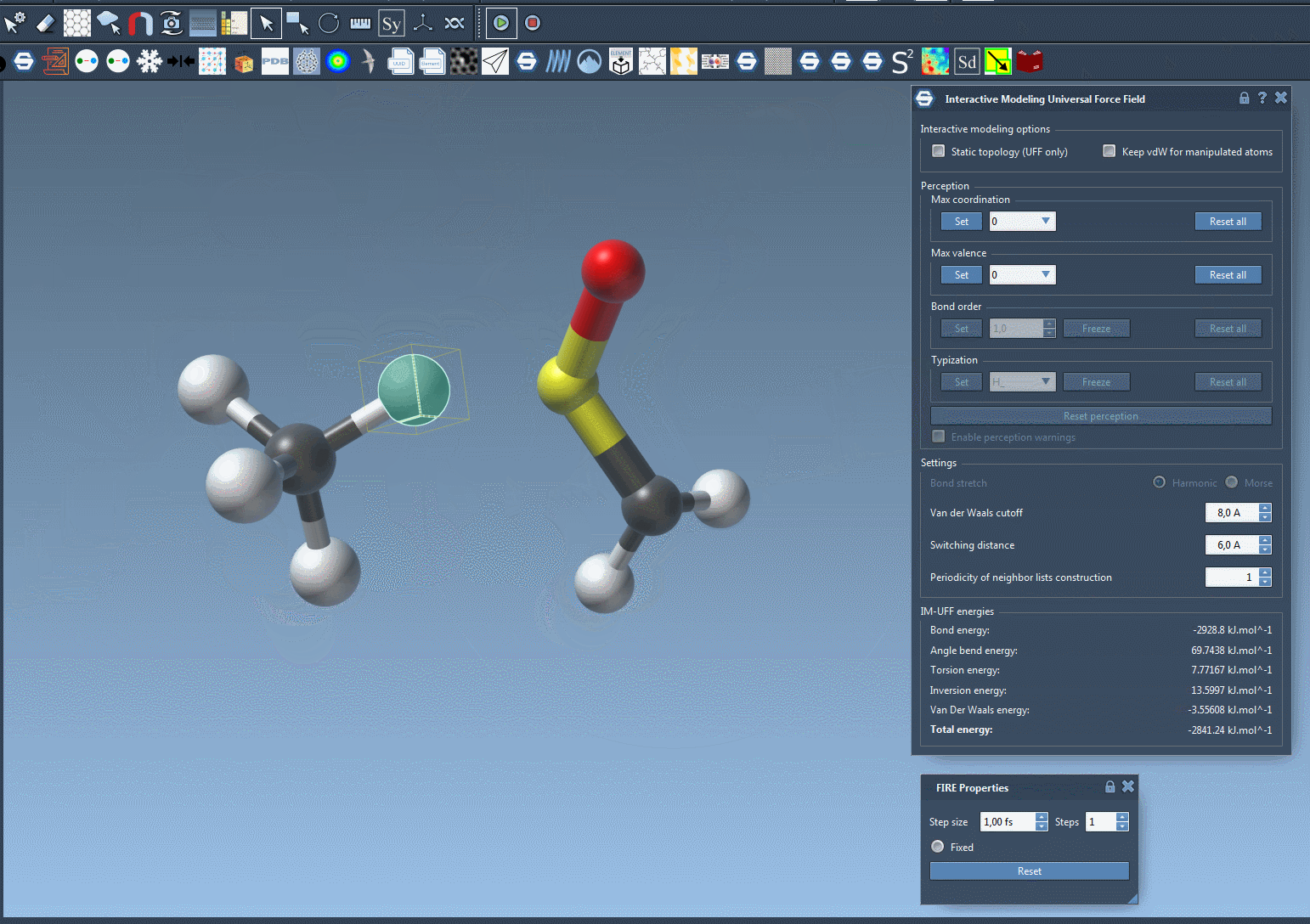
Customizing UFF and IM-UFF
The IM-UFF interaction model allows one to set the van der Waals cutoff, the van der Waals switching distance, and the periodicity of the neighbor list construction for the two simulation modes: UFF only and IM-UFF. For details about these parameters, see our previous post describing the UFF interaction model.
The IM-UFF interaction model also offers several options to customize the automatic typization in UFF/IM-UFF. The description of these options can ones again be found in our previous post describing the UFF interaction model. However, note that when IM-UFF is running (ie. when the static topology is unchecked), only the maximum coordination and maximum valence can be set. In this mode, the bond order and typization cannot be directly set since they take continuous values in function of the current positions of the atoms, to allow smooth transitions between different topologies. For more details, see our reference article [3].
Finally, atoms can also be deleted from the model and added to the model “on the fly” i.e. the typization is then updated automatically as in the UFF interaction model. For more details, see once again our previous post describing the UFF interaction model.
If you have any questions or feedback, please use the SAMSON forum.
[1] A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. Goddard III and W. M. Skiff. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. Journal of the American Chemical Society 114.25 (1992): 10024-10035. [2] S. Artemova, L. Jaillet and S. Redon. Automatic molecular structure perception for the universal force field. J. Comput. Chem. 2016, 37, 1191-1205. [3] Jaillet, Léonard, Svetlana Artemova, and Stephane Redon. IM-UFF: extending the Universal Force Field for interactive molecular modeling. Journal of Molecular Graphics and Modelling, 77 (2017): 350-362.