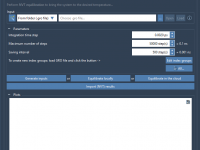
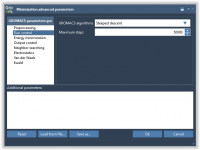
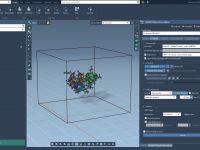
GROMACS Wizard – Computing in the cloud
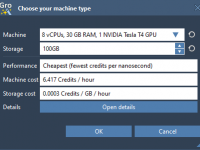
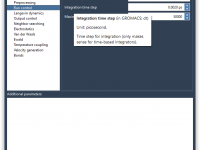
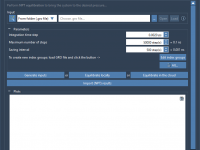
This section is part of the GROMACS Wizard tutorial. GROMACS Wizard allows you to launch GROMACS computations for NVT Equilibration, NPT Equilibration, Production Molecular Dynamics Simulation steps in the Cloud in just a few clicks. In this section, you will learn how to launch GROMACS computations in the Cloud from GROMACS Wizard on an example of the NVT Equilibration step. This procedure is the same for NPT Equilibration and Production Molecular Dynamics Simulation steps. Prerequisites Before going further, please first read…