GROMACS Wizard – Batch Computations
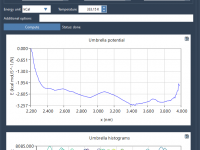
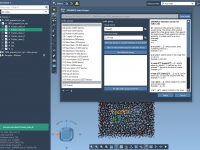
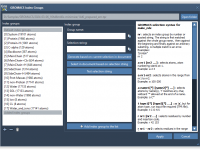
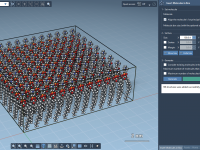
This section is a part of the GROMACS Wizard tutorial. This tutorial demonstrates how to perform batch computations using GROMACS Wizard for a set of initial conformations of a single molecular system. To use an existing trajectory, load it in SAMSON. You can also create a trajectory or a set of conformation in SAMSON using various tools: editors (Move editors, Twister), various extensions (AutoDock Vina Extended, Normal Modes Analysis, Ligand Path Finder, Protein Path Finder, protein-protein docking with Hex, etc.), animations (Dock,…