Explore ligand strain with Strain Explorer in SAMSON#
Use Strain Explorer to analyze how much a ligand relaxes from its starting conformation, compare local and global strain estimates, inspect a 2D strain map, and export results for reporting or further work in SAMSON.
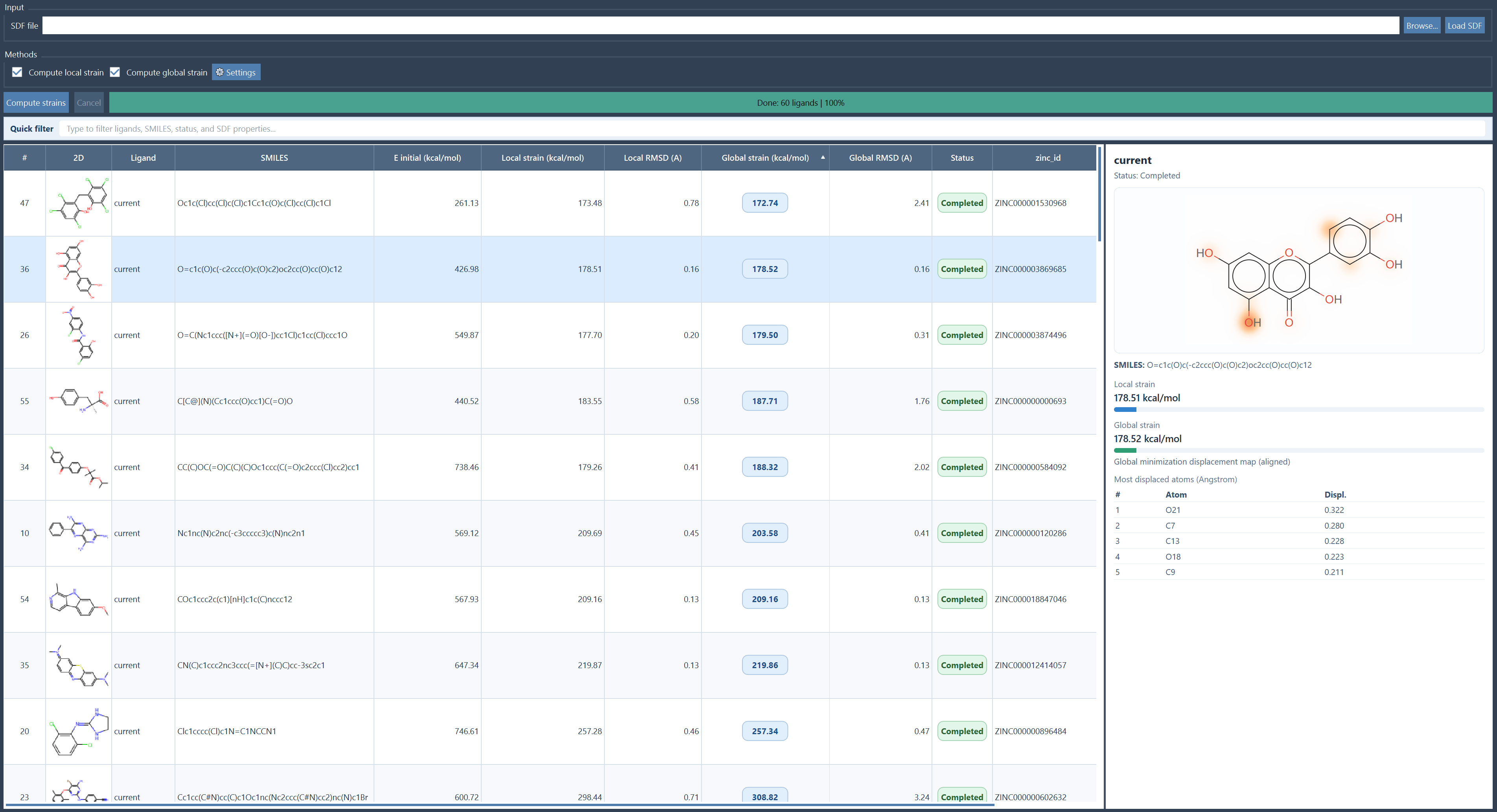
Strain Explorer is designed for ligand libraries stored in SDF files. It keeps the imported ligands in memory, computes strain values with either UFF or UMA backends, and displays the results in a sortable table with 2D depictions, SMILES, SDF properties, strain values, RMSDs, and status messages.
What Strain Explorer does#
Strain Explorer can help you:
- load one or many ligands from an SDF file;
- compute local strain from the imported pose;
- compute global strain using a torsional search over rotatable bonds;
- compare ligands in a table with sorting and quick filtering;
- visualize a ligand-specific strain map in a side panel;
- send selected ligands to the SAMSON document as aligned conformations;
- export selected or all ligands to SDF;
- export a ligand report to PDF.
Before you start#
- Add the Strain Explorer SAMSON Extension.
- Make sure the required computation backend is available:
- UFF for the default workflow. See Universal Force Field.
- UMA if you want to use the UMA backend and have it installed. See UMA Force Field.
- Prepare an SDF file containing one or more ligands.
Input requirements
Strain Explorer supports V2000 and V3000 SDF files and preserves the SDF properties as table columns.
Each ligand must contain a SMILES property. Strain Explorer uses it for the 2D depiction and the strain panel. If a ligand is missing SMILES, loading is aborted.
Use 3D coordinates when possible
Strain is computed from the imported coordinates. In practice, 3D ligand conformations are the most meaningful input. If you import flat 2D coordinates, the computed strain mostly reflects how far that drawing is from a relaxed 3D geometry.
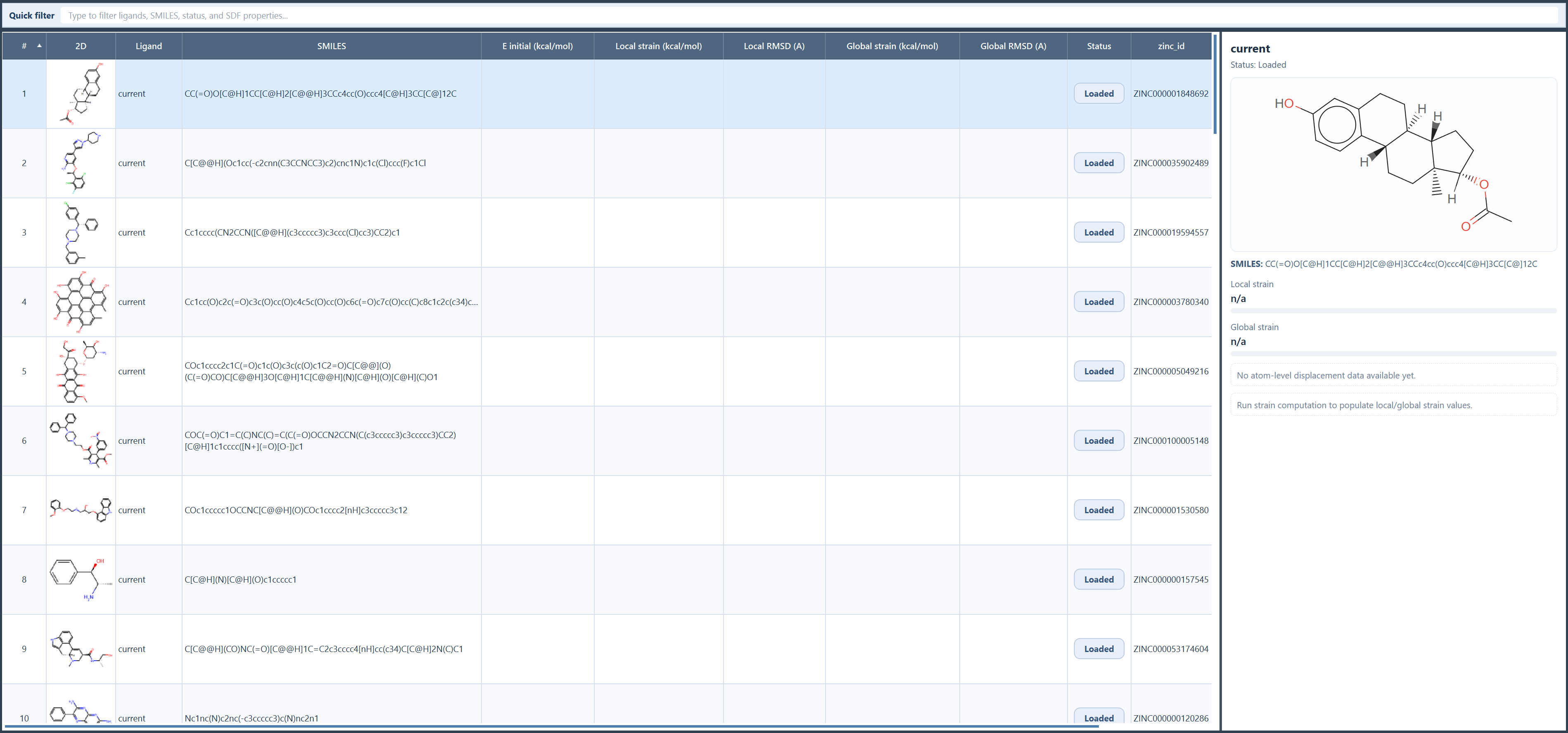
Open the app and load an SDF file#
Open Strain Explorer from Home > Apps > Biology or by using Find everything....
Then:
- Click Browse... and select your SDF file.
- Click Load SDF.
After loading:
- the ligands appear in the results table;
- the table includes the ligand name, SMILES, 2D depiction, computed metrics, status, and all imported SDF properties;
- the Quick filter lets you search across all visible columns;
- the ligands are not added to the SAMSON document yet.
Understand the two strain calculations#
Strain Explorer reports how much the ligand energy decreases when the imported conformation is relaxed.
- Local strain =
E(initial) - E(local minimum) - Global strain =
E(initial) - E(global minimum estimate)
Higher strain usually means the starting conformation stores more conformational tension and relaxes more strongly during optimization.
Local strain#
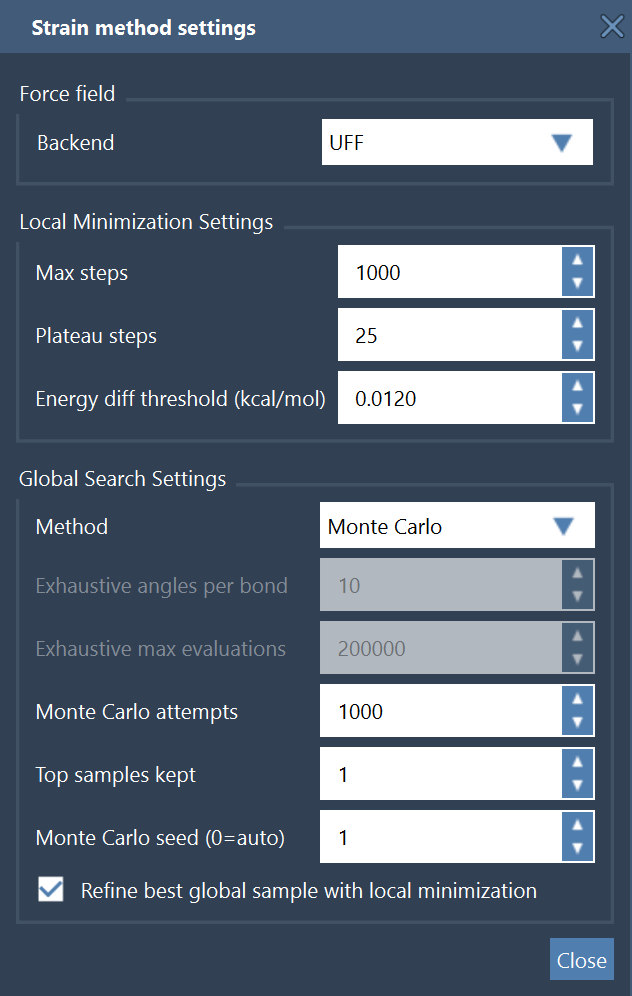
The local calculation starts from the imported ligand coordinates and runs a local minimization with the selected force field.
You can adjust:
- Max steps
- Plateau steps
- Energy diff threshold
The minimization stops when one of these conditions is met:
- the maximum number of steps is reached;
- the energy change remains below the chosen threshold for enough consecutive steps.
This is useful when you want to know whether the imported ligand is already close to a nearby minimum.
Global strain#
The global calculation tries to find a lower-energy conformer that may not be reachable by a purely local relaxation.
Strain Explorer does this in several stages:
- It detects rotatable single bonds.
- It builds an articulated rigid-body model of the ligand.
- It samples torsion angles using either: - Exhaustive search, or - Monte Carlo search.
- It keeps the lowest-energy sampled conformers.
- It can optionally refine the best sampled conformer with a local minimization.
Important
The global result is a global minimum estimate, not a mathematical guarantee of the absolute global minimum. The quality of the estimate depends on the search mode and the chosen sampling settings.
If a ligand has no rotatable bonds, the global stage becomes trivial and optional refinement is usually all that remains.
Exhaustive vs Monte Carlo#
Use Exhaustive when the ligand has only a limited number of rotatable bonds and you want systematic sampling.
Use Monte Carlo when the ligand is more flexible and exhaustive sampling would become too expensive.
The main global settings are:
- Exhaustive angles per bond: how finely each torsion is discretized;
- Exhaustive max evaluations: safety cap on the total number of scored conformers;
- Monte Carlo attempts: number of random torsional samples;
- Monte Carlo seed: fixed seed for reproducible random sampling;
- Top samples kept: number of low-energy candidates retained during the search;
- Refine best global sample with local minimization: performs a final local relaxation of the best sampled pose.
Run the computation#
Once your ligands are loaded:
- Select Compute local strain, Compute global strain, or both.
- Open Settings if you want to change the force field or the local/global parameters.
- Click Compute strains.
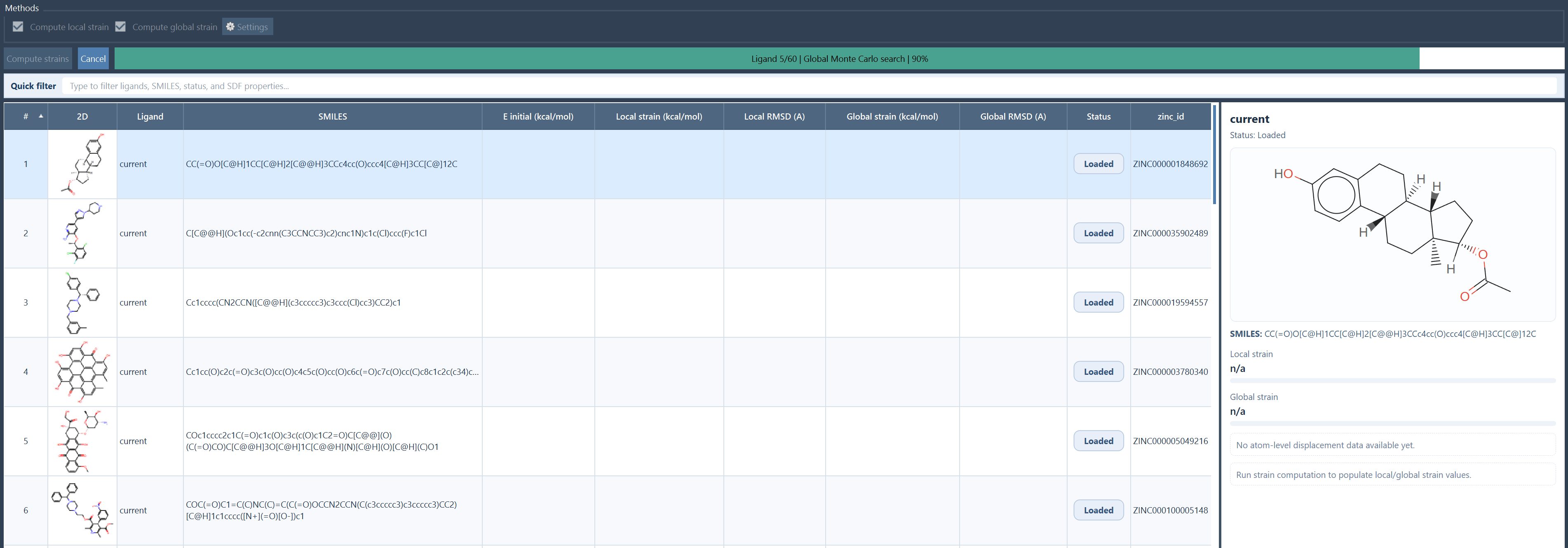
During the run:
- the progress bar reports the current ligand and stage;
- Cancel stops the run cooperatively;
- errors on one ligand do not necessarily stop the whole batch;
- each ligand receives its own status such as Completed, Completed with warnings, Cancelled, or Error.
Read the results table#
After the run, the table becomes the main comparison view.
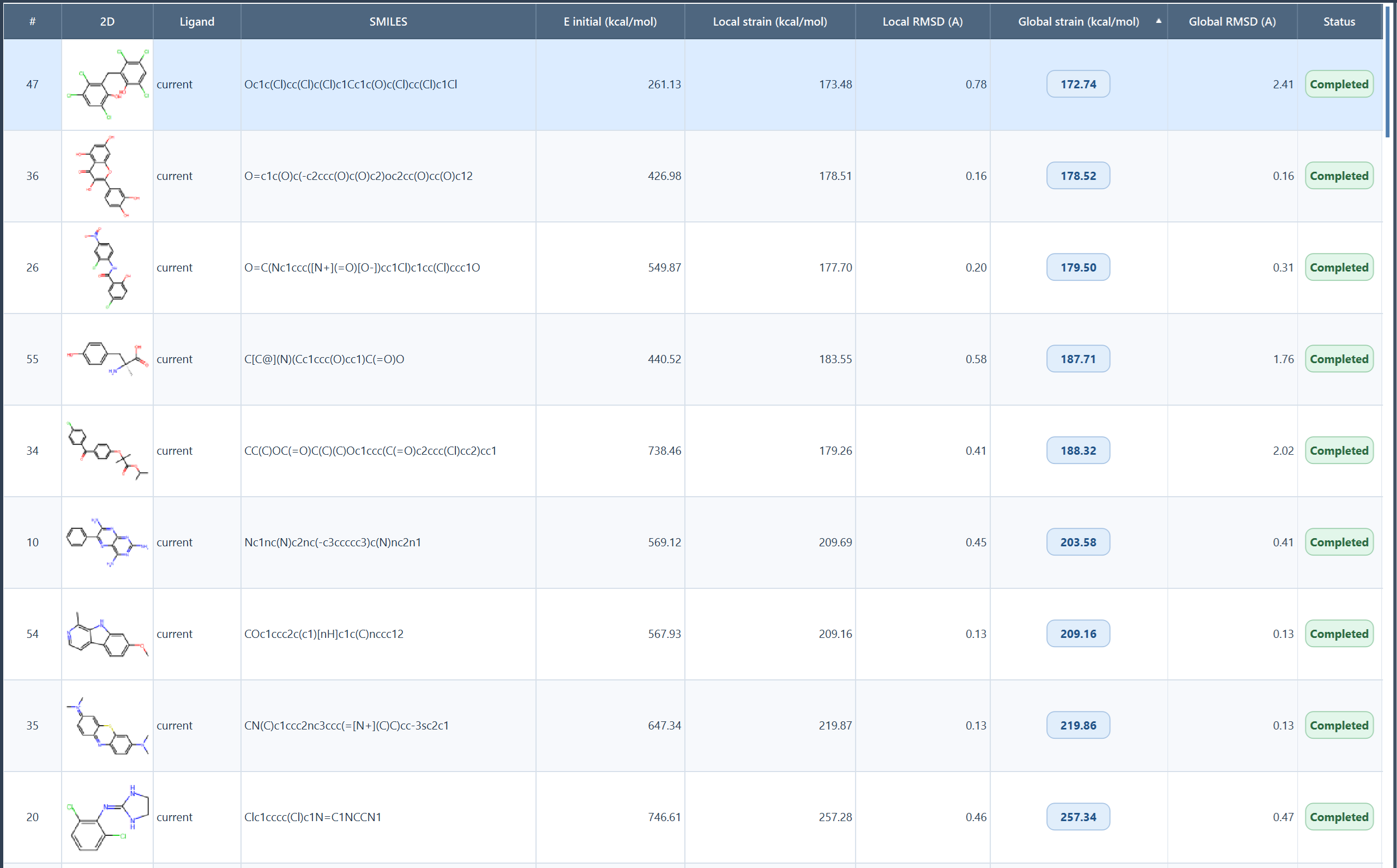
The most important columns are:
- E initial (kcal/mol): force-field energy of the imported conformation;
- Local strain (kcal/mol): energy released by local minimization;
- Local RMSD (A): structural change after local minimization, after rigid alignment;
- Global strain (kcal/mol): energy released relative to the best global conformer found;
- Global RMSD (A): structural change after the global stage, after rigid alignment;
- Status: summary of success, warnings, or failure for that ligand.
You can:
- sort numerically by strain or RMSD;
- filter by ligand name, SMILES, status, or any SDF property;
- select one or more rows for visualization or export.
How to interpret local vs global strain
- If local strain and global strain are both small, the imported conformation is already close to a relaxed state.
- If global strain is much larger than local strain, the ligand likely has a lower-energy torsional arrangement outside the local basin.
- If local strain is already large and global strain is similar, the ligand may simply need local relaxation rather than a fundamentally different torsional state.
Use the strain panel#
When you select a row, the right-hand panel shows a ligand summary with:
- a 2D depiction generated from the ligand SMILES;
- local and global strain bars;
- a ranked list of the most displaced atoms;
- a color overlay on the 2D depiction.
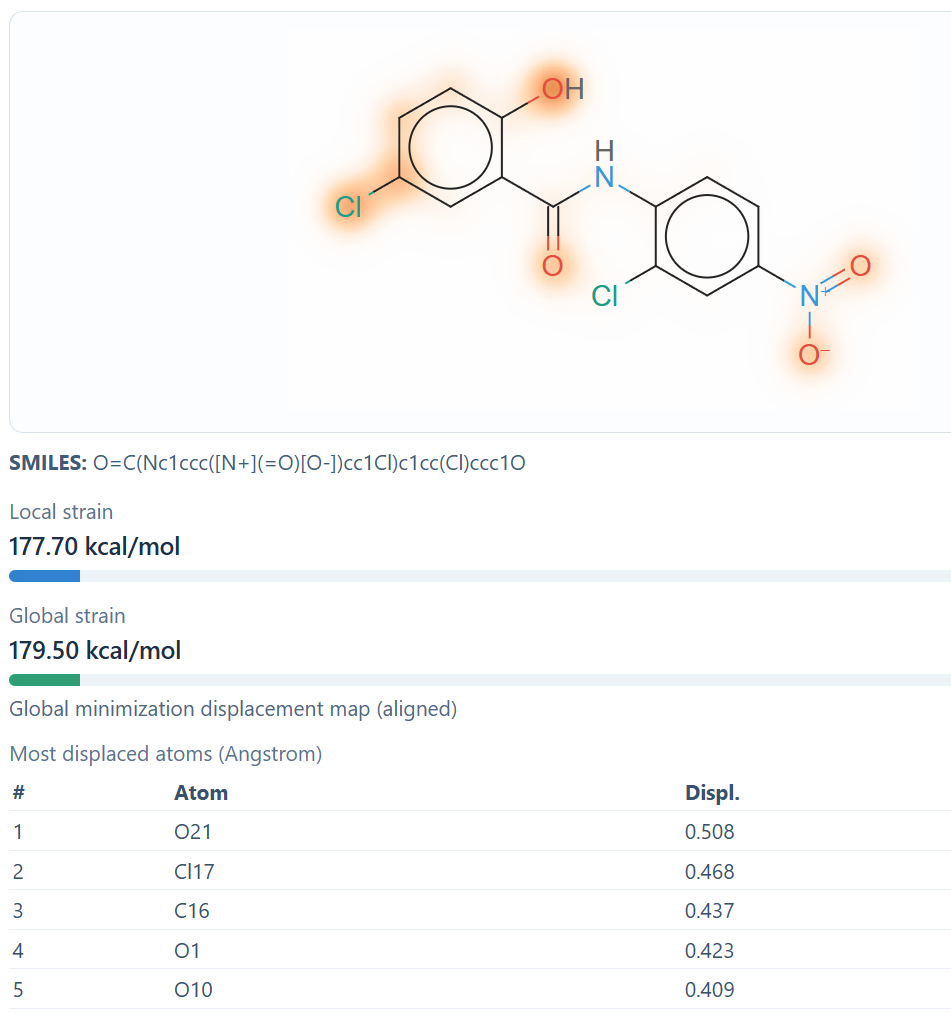
What the strain map represents
The current strain map is a displacement-based visualization. It highlights how much atoms move between the original conformation and the minimized conformation after alignment.
If global results are available, the panel uses the global minimized structure. Otherwise, it falls back to the local minimized structure.
This makes the panel very useful for spotting where the ligand geometry changes the most, for example around flexible substituents or strained ring-adjacent atoms.
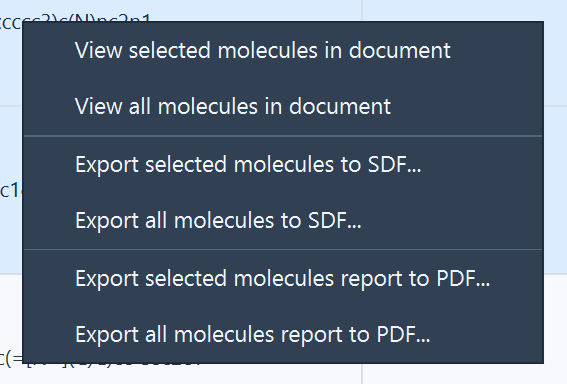
View ligands in the SAMSON document#
Right-click in the results table to open the context menu, then choose:
- View selected molecules in document
- View all molecules in document
Strain Explorer adds one folder per ligand to the active document and creates aligned structural models for:
- the original imported conformation;
- the locally minimized conformation;
- the globally minimized conformation.
This is useful when you want to compare conformers directly in 3D, add visual models, or continue analysis with other SAMSON tools.
Export results#
Export to SDF#
From the same context menu, you can export the selected or all ligands to SDF.
The exported file contains:
- the ligand structure;
- the original imported coordinates;
- the original SDF properties;
- computed properties such as Local strain, Local RMSD, Global strain, and Global RMSD when available.
This is convenient for downstream filtering or for sharing annotated ligand libraries.
Export to PDF#
You can also export a PDF report for the selected or all ligands.

The report includes:
- a cover summary for the exported ligand set;
- counts of local and global successes;
- the ligand with the lowest strain in the exported batch;
- one card per ligand with a depiction, strain values, RMSDs, and SMILES.
This is useful for medicinal-chemistry review meetings or quick project reports.
Practical tips#
- Start with local strain only if you want a fast first pass.
- Use Monte Carlo for flexible ligands with many rotatable bonds.
- Use Exhaustive only when the torsional search space remains manageable.
- Keep global refinement enabled if you want the best sampled conformer to be relaxed before reporting strain.
- Use the Quick filter to narrow a large library by status, ID, scaffold, or assay property.
Current limitations to keep in mind#
- Input is currently SDF only.
- A SMILES property is required for each ligand.
- The strain panel is currently displacement-based, not a full per-atom energy decomposition.
- Global strain is a search-based estimate and depends on sampling quality.
That is all you need to start using Strain Explorer for ligand strain analysis in SAMSON.
Need Help?#
Have questions or feedback? Feel free to reach out via the Forum, via e-mail, via the Feedback button in SAMSON, or by directly discussing with us.