Dock ligands and libraries of ligands with AutoDock Vina Extended#
Use AutoDock Vina Extended SAMSON Extension to dock one ligand, a ligand library, or several receptors in SAMSON, then inspect and analyze the resulting poses in the same workspace.
The AutoDock Vina Extended SAMSON Extension wraps the popular protein-ligand docking program AutoDock Vina 1 and extends its functionality by providing the possibility to:
- easily set up the system (a receptor or a receptor library, a ligand or a ligand library), including flexible side chains, rotatable bonds, and types of locked bonds;
- easily set up the search domain based on the receptor, a ligand, or a binding site, or by manually adjusting it in the Viewport or in the interface;
- perform the minimization of ligands;
- specify multiple advanced parameters;
- perform docking, re-scoring, or generate the project to run it on your cluster;
- filter the results;
- save the results, including automatic export of each mode into a separate MOL2 file;
- analyze the results, export the results table and plots;
- load the previously computed results.
What you will learn#
In this tutorial, you will learn how to set up receptors and ligands, run docking with AutoDock Vina Extended in SAMSON, and inspect the results.
Before you start#
- Add AutoDock Vina Extended SAMSON Extension.
- Download the 2AZ8 tutorial archive.
- If your own system is not already clean, be ready to prepare it before docking.
First steps#
Launch SAMSON and open the 2AZ8-tutorial.sam file provided in the archive. It will open structural models of a protein (2AZ8-A and 2AZ8-B) and of a ligand (2AZ8-IA) bound to the protein - for the sake of the tutorial, the protein is put in the Receptor folder and the ligand is put in the Ligand folder.
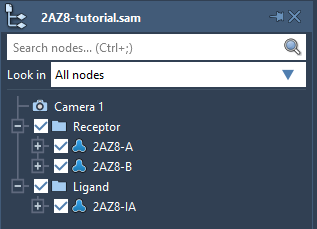
Tip
If you don't see the Document view, you can enable it in the Interface menu or via Ctrl/Cmd + 1.
Preparation of the system#
We need to prepare the system first:
- Remove atoms with alternate locations.
- Remove water and monatomic ions. This step is optional since water and monatomic ions will not be considered by AutoDock Vina Extended.
- Remove ligands, co-factors, and other unnecessary small molecules present in the system.
- Add hydrogens, if the system does not have them set. AutoDock Vina needs polar hydrogens to determine hydrogen bonds (H-bonds). Note that if the system has hydrogens added based on some protonation you should not modify hydrogens.
To do all of this in a single step, go to Home > Prepare and check the above-mentioned options as shown in the image below.

If your system has missing residues or heavy atoms (e.g., missing atoms in side chains), you can also use the PDBFixer extension to fix a protein. This extension can also be used to add hydrogens for specific pH.
See also
Please refer to the Protein Preparation & Validation tutorial for more information on how to prepare and fix protein systems.
Note
The system in the tutorial file has all the water already removed and hydrogens already added.
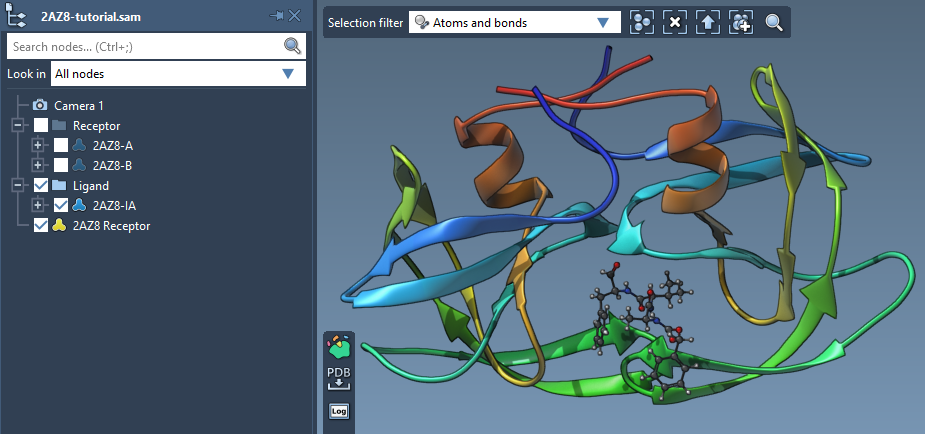
Tip: visualization
If there is no secondary structure shown in the Viewport, you can add it via Visualization > Visual model > Ribbons or Visualization > Visualize > Protein-ligand.
You can also hide/show the atomistic representation by toggling the boxes in front of the protein structure in the Document view.
To learn more about visual models in SAMSON please follow interactive tutorials in SAMSON (Help > Tutorials) or the User guide: Visualizing tutorial.
Setup of the system#
Let's now open the AutoDock Vina Extended app by clicking on it in the Home > Apps > Biology menu. You can also find it using the Find everything... search box (Shift+E) in the top menu of SAMSON - just start typing the name.
Click on New docking project.
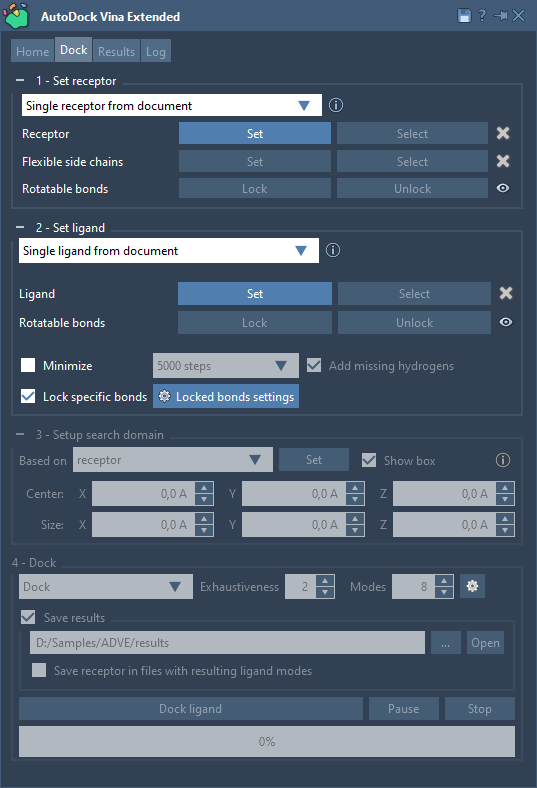
Now we need to set up the system:
- a receptor or a receptor library;
- [optional] receptor's flexible side chains in case of a single receptor;
- a ligand or a ligand library.
Setup of the receptor#
Select the receptor - 2AZ8-A and 2AZ8-B structural models - in the Document view and then click on the Set button in the Set receptor part. You can also use Select > Biology > Receptors to automatically select receptors. You will see the search domain set based on the receptor, with its box shown in yellow in the Viewport.
If you want to unset the receptor then just clear the selection and click on the Set button.
Setup of the receptor's flexible side chains#
The setting of flexible side chains allows performing docking to a relatively flexible receptor with rotatable side chains. Note that having flexible side chains may result in more realistic docking but significantly increases the docking time.
For simplicity, in this tutorial, we will not be specifying flexible side chains. But below you can find how to set up the receptor's flexible side chains.
You can specify the receptor's flexible side chains using one of the following ways:
-
Select them in the Document view.
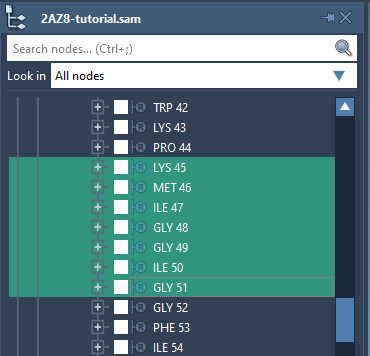
-
Select them in the Viewport by switching the Selection filter in the top-left corner of the Viewport to Residues and selecting residues one by one using Ctrl and Alt buttons to add/remove to the current selection. You will need to have the structural model visible in the Viewport.
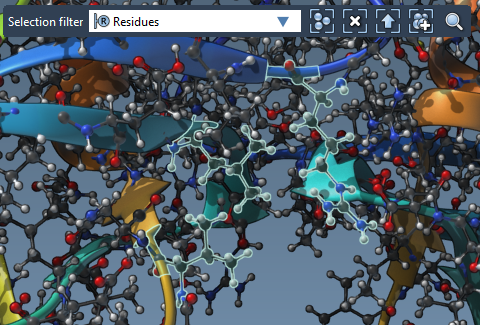
Tip
See the User Guide: Selecting or interactive tutorials in SAMSON.
-
If you have a bound ligand and you want to specify residues around it as flexible, then select this ligand and go to Select > Biology > Binding sites and specify parameters as necessary and click OK:
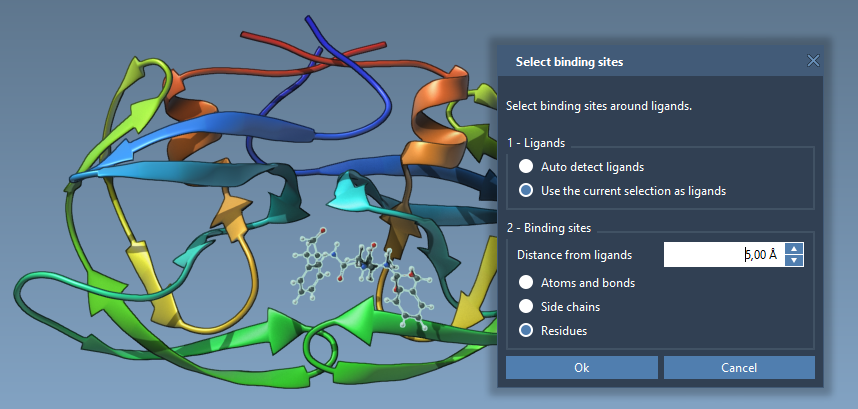
Once residues with flexible side chains are selected, click on the associated Set button in the Set receptor part.
Once you set up flexible side chains, you will see in the Viewport the rotatable bond controllers represented by green cylinders superimposed with bonds. By clicking on them, you can switch the bonds from rotatable to not rotatable and back (green - rotatable, red - not rotatable). See more about it in the Setup of rotatable bonds section.
Setup of a ligand or a ligand library#
The AutoDock Vina Extended allows you to dock either a ligand or a library of ligands.
Setup of the ligand#
Let's first see how to set up a ligand. First, check the Single ligand in the Set ligand part, then select the ligand - 2AZ8-IA - in the Document view and click on the associated Set button in the Set ligand part.
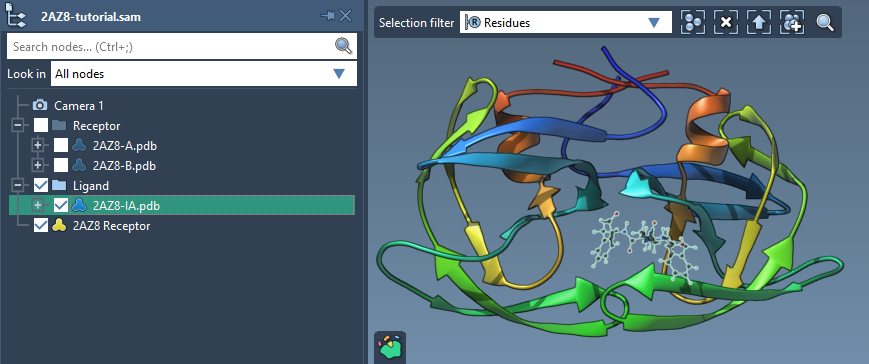
Setup of rotatable bonds#
Once you set up a ligand, you should see in the Viewport the rotatable bond controllers represented by green cylinders superimposed with bonds. Zoom in on the ligand (select the ligand in the Document view and then press Shift+Space).
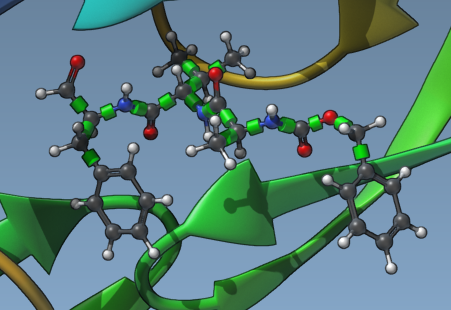
By clicking on these controllers, you can activate or deactivate these rotatable bonds - switch the bonds from rotatable to not rotatable and back - they will change colors when you switch their state. Green means the bond can rotate, and red means the bond cannot rotate. Note that having rotatable bonds may result in more realistic docking but increases the docking time.
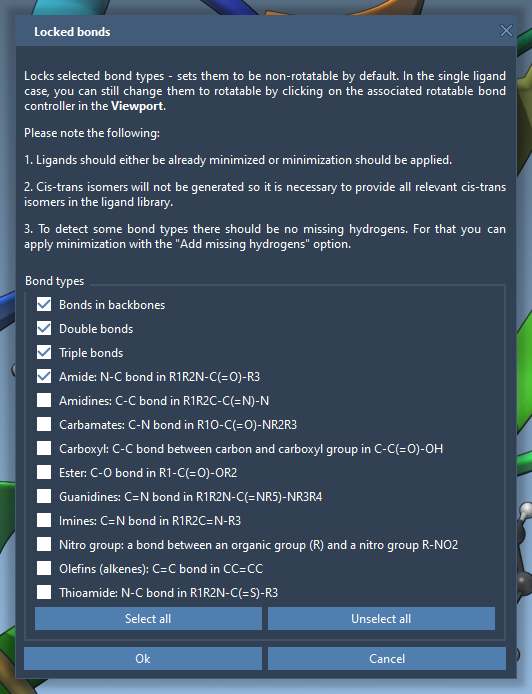
You can also use the option to lock specific bond types (make them non-rotatable). For this, click on the Locked bonds settings and check the types of bonds that you want to be non-rotatable (locked) and click OK.

Then check the Lock specific ligand bonds option.
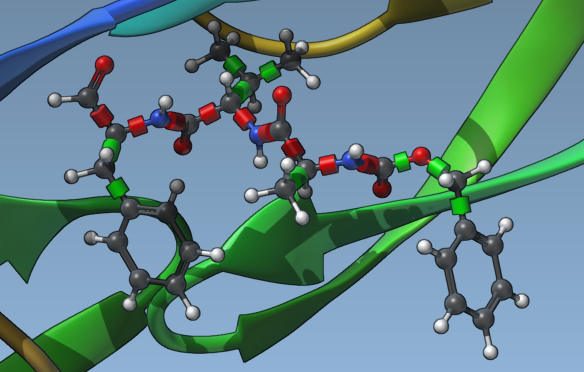
Note
- Ligands should either be already minimized or minimization should be applied (see the Minimization of ligands section below). Locking of bonds does not affect minimization i.e., the locked bonds will be rotatable during minimization.
- Cis-trans isomers will not be generated so it is necessary to provide all relevant cis-trans isomers in the ligand library.
Setup of the ligand library#
In this tutorial, we will be docking not a single ligand but a ligand library, which is provided in the same archive. It is a small sample library with some random ligands obtained from the ZINC library.
In the library, you can provide files of different formats (e.g., MOL2, SDF) and multiple ligands per file.
To dock a ligand library, check the Ligand library option and click on the  button to browse the directory with the ligand library.
button to browse the directory with the ligand library.
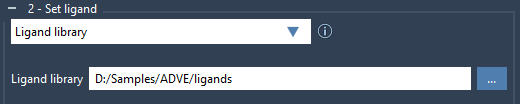
Note that by default all the ligands in the library will be considered fully flexible (meaning that all the rotatable bonds will be active). You can lock specific bond types thanks to the aforementioned Lock specific ligand bonds option (first, verify the Locked bonds settings). Please see the Setup of the rotatable bonds section.
Minimization of ligands#
If you want to minimize ligands before docking, which might be necessary in some cases, for example, if ligands in the library are in 2D, then check the Minimize option and choose the preset (the maximum number of minimization steps and stopping criteria). Check the Add missing hydrogens option if ligands do not have hydrogens already set - hydrogens are necessary for detecting bond types during the minimization and polar hydrogens are necessary for docking.

The minimization is stopped based on some stopping criteria - when either the energy difference between steps is less than some threshold during at least some consecutive minimization steps or the maximum number of minimization steps has been reached. These values are determined by the minimization preset.
Setup of the search domain#
The next step is to set up the search domain. By default, the search domain is determined once you set the receptor based on the whole receptor. If you already know the binding site, you can specify the search domain that surrounds it - this will not only limit the search domain but will also lead to performing fewer computations decreasing the overall docking time.
Tip
If you want to hide/show the search domain in the Viewport, then toggle the Show box option.
You can set up the search domain using one of the following ways:
- Modify the center and the size of the domain in the interface.
- Adjust the search domain box directly in the Viewport thanks to the controllers placed in the corners of the yellow search box visible in the Viewport - just press on the sphere in the corner of the box and drag it to modify its position.
-
Set it up based on the current selection: select nodes and choose the Based on pocket/selection option from the list and click Set.

-
Set it up based on the bound ligand/binding pocket: select the bound ligand and go to the Select > Biology > Binding sites, after the binding site is selected choose the Based on pocket/selection option from the list and click Set. See the example below.
You can always just set the search domain directly in the Viewport thanks to the search box controllers.
Let's set up the search domain based on the binding site of the 2AZ8-IA bound ligand. Select the 2AZ8-IA ligand in the Document view and go to Select > Biology > Binding sites and specify parameters as shown in the image below and click OK:
This should select residues around the ligand. Now choose the Based on pocket/selection option from the list and click Set.
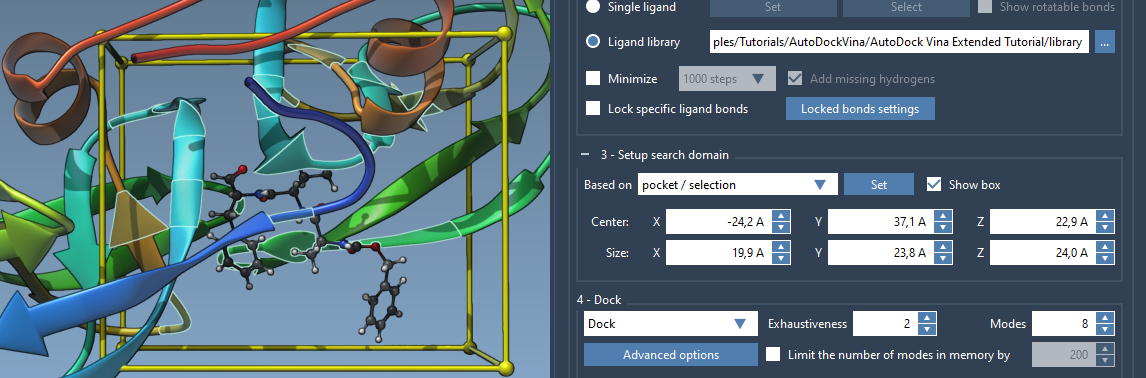
Please note that the search domain is set in the global XYZ coordinates. If you want, you can always rotate or align the system using the Move editors to better align the binding site in the global XYZ coordinates.
Running the docking and more#
The next step is to run the docking!
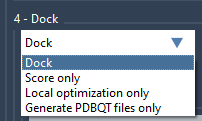
The Dock part provides the following options:
- Dock - performs docking.
- Score only - performs scoring of ligands in their current positions without minimization.
- Local optimization only - performs local optimization only and scores ligands in their current positions.
- Generate PDBQT files only - generates a project with PDBQT files for the receptor and ligands, and a configuration file - you can use these files to launch Vina computations on your own cluster. If the minimization option has been checked, the ligands will be minimized before generating PDBQT files. In the case of the use of a ligand library, the PDBQT files for ligands will be generated with keeping the same hierarchy as in the ligand library.
Choose the Dock from the list since we want to perform the docking.

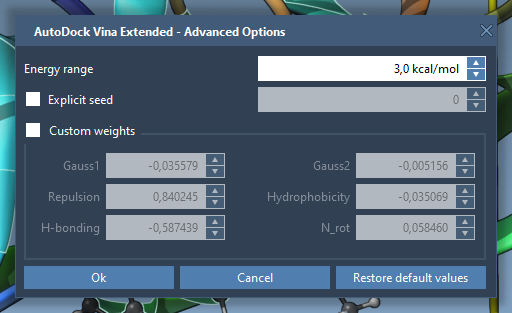
To change exhaustiveness, the maximum number of modes, or other parameters click on Options:
- Set the Exhaustiveness parameter (the higher the better, but the longer the search) to 8.
- Set the Max. number of generated binding modes parameter (the maximum number of modes returned by the search algorithm) to 20.
You can also modify other parameters, e.g., scoring function (vina or vinardo), the energy range for the resulting modes, grid spacing, weights of scoring functions, etc. We do not advise modifying the weights; if you modified them, you can always restore their default values by clicking on the Restore default values button.
If you want your results to be automatically saved on disk, then check the Save results option and select the directory where you want your results to be saved. This will create a project directory with a timestamp in its name; this directory will contain the receptor and ligand files, generated PDBQT files, docked PDBQT files, and MOL2 files for each ligand's mode. You can later load these results back to AutoDock Vina Extended (see the Loading previously computed results section). Please note that you shouldn't modify the hierarchy of the project directory or remove files if you want to be able to load them back into AutoDock Vina Extended.
Please check the Save results option and select the directory where you want your results to be saved. We will need these saved results in the Loading previously computed results section.
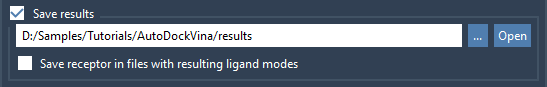
Now, press the Dock library button and wait for the results - it should take a couple of minutes. There might be some pop-up dialogs asking for options to import ligands from the library folder. If you are docking a ligand library, you can always pause, resume, or stop the computations - the pause and stop will be applied only after the computations for the current ligand are finished.
Visualizing results#
As soon as the computations are done, a new folder with the results should be added to the document containing the following:
- A note called
Configurationwith the docking parameters. If you want to see the note with the docking parameters, right-click on it and choose Inspect in the context menu. - A subfolder called
Top ligandswith structural models of the top binding ligands (with the best ranked pose already displayed). - Top conformations of the top ligands allow you to display poses of these ligands.
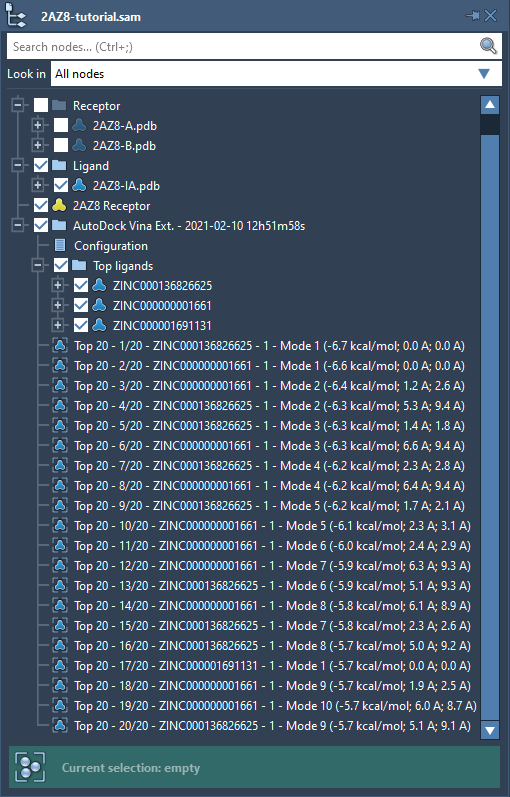
To restore a conformation, either double-click on it in the Document view or right-click on it and in the context menu choose Restore conformation.
You can also create structures (structural models) from conformations, which makes it easier to see clusters of conformations. To achieve this, right-click on one or more conformations and choose Create structural models from conformations. This will create structural models for all selected poses, so you can see where clusters are.
Note that you can hide structural models from displaying in the Viewport by unchecking them in the document.
Note that all the conformations are stored in a directory with results if you choose to save the results, or if you loaded the previously computed results. You can also export the selection, for example as MOL2 or PDB files, by clicking on Home > File > Save selection as... (Ctrl/Cmd + E). To export the ligand's conformation in a separate file, restore this conformation by double-clicking on it and then select the ligand's structural model and click Home > File > Save selection as....
The Results tab in the AutoDock Vina Extended allows you to see the results table and plot, filter the results based on some parameters (e.g., name, affinity, RMSD, etc.), export the results table in a CSV file, export the affinity vs compounds plot as an image or in a CSV file. There is a horizontal splitter line between the results table and the plot that you can drag to modify the size of the results table and the plot.
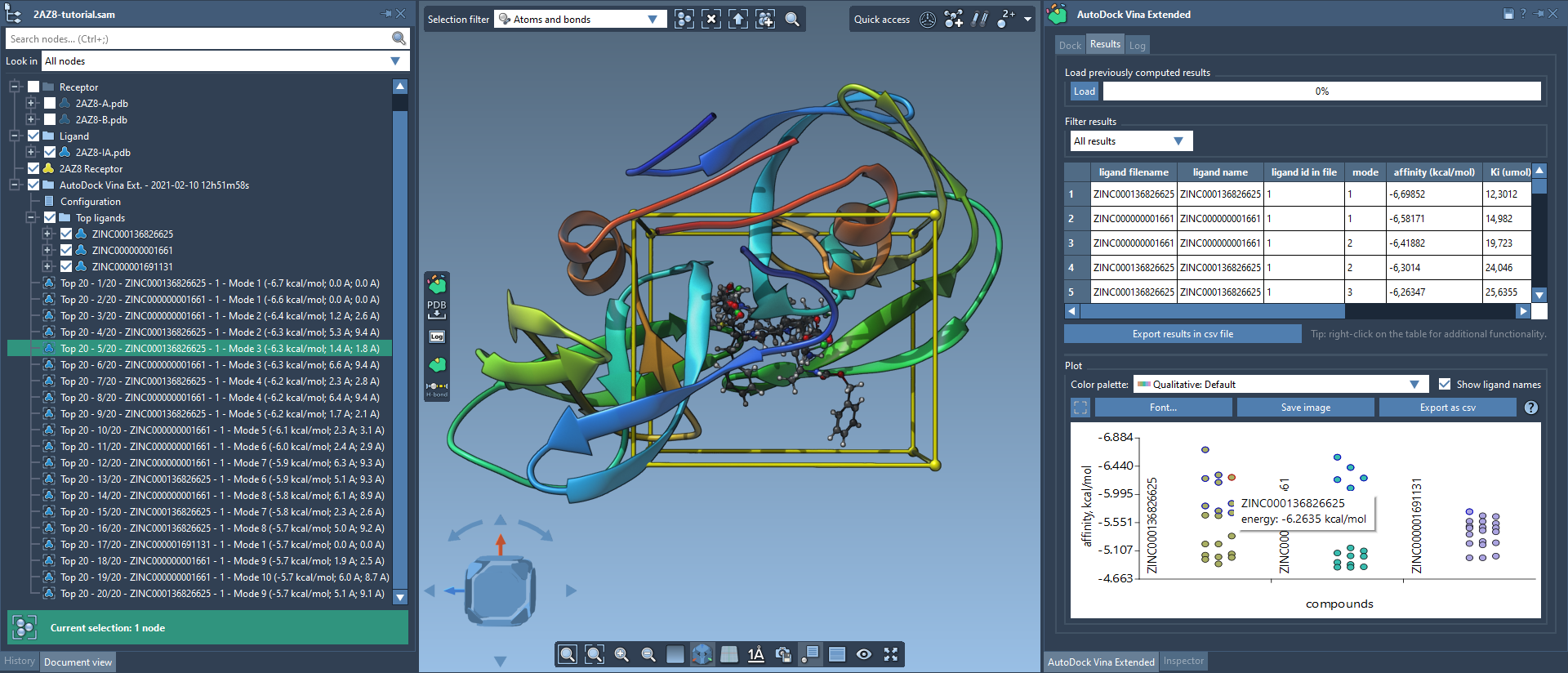
The results table allows you to sort by columns, copy the data, select ligands and conformations, restore conformations, and export ligands and conformations in the document that are not present among the top ligands and conformations. To do that, right-click on a row and choose a corresponding action from the context menu.
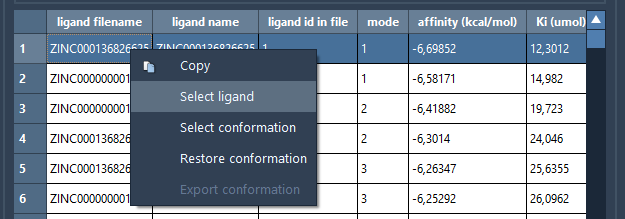
The plot allows for selecting and restoring conformations. By clicking on a data point corresponding to a conformation present in the document, this conformation will be selected in the Document view and vice versa. By double-clicking on a data point, the corresponding conformation, if present in the document, will be restored.
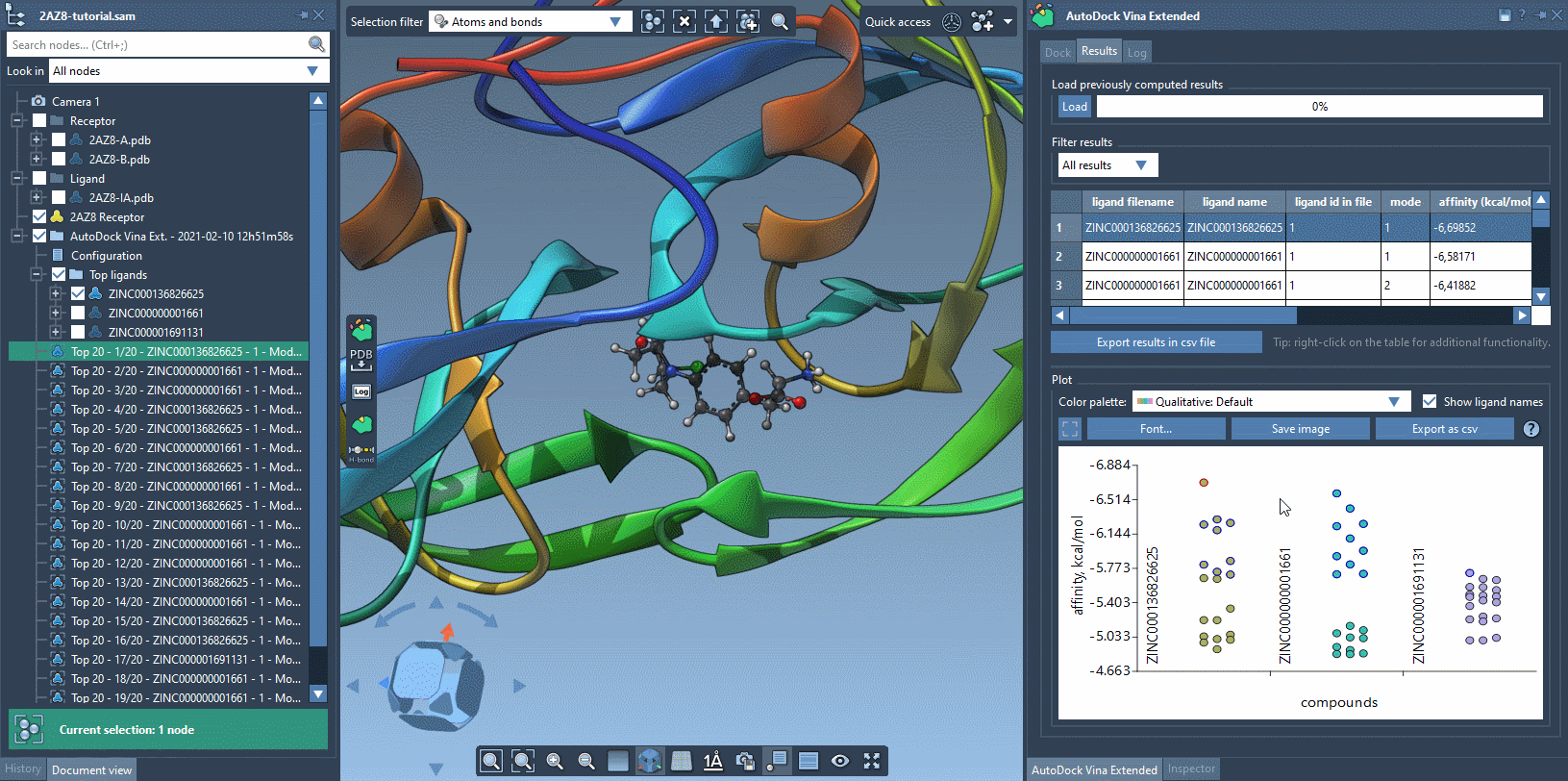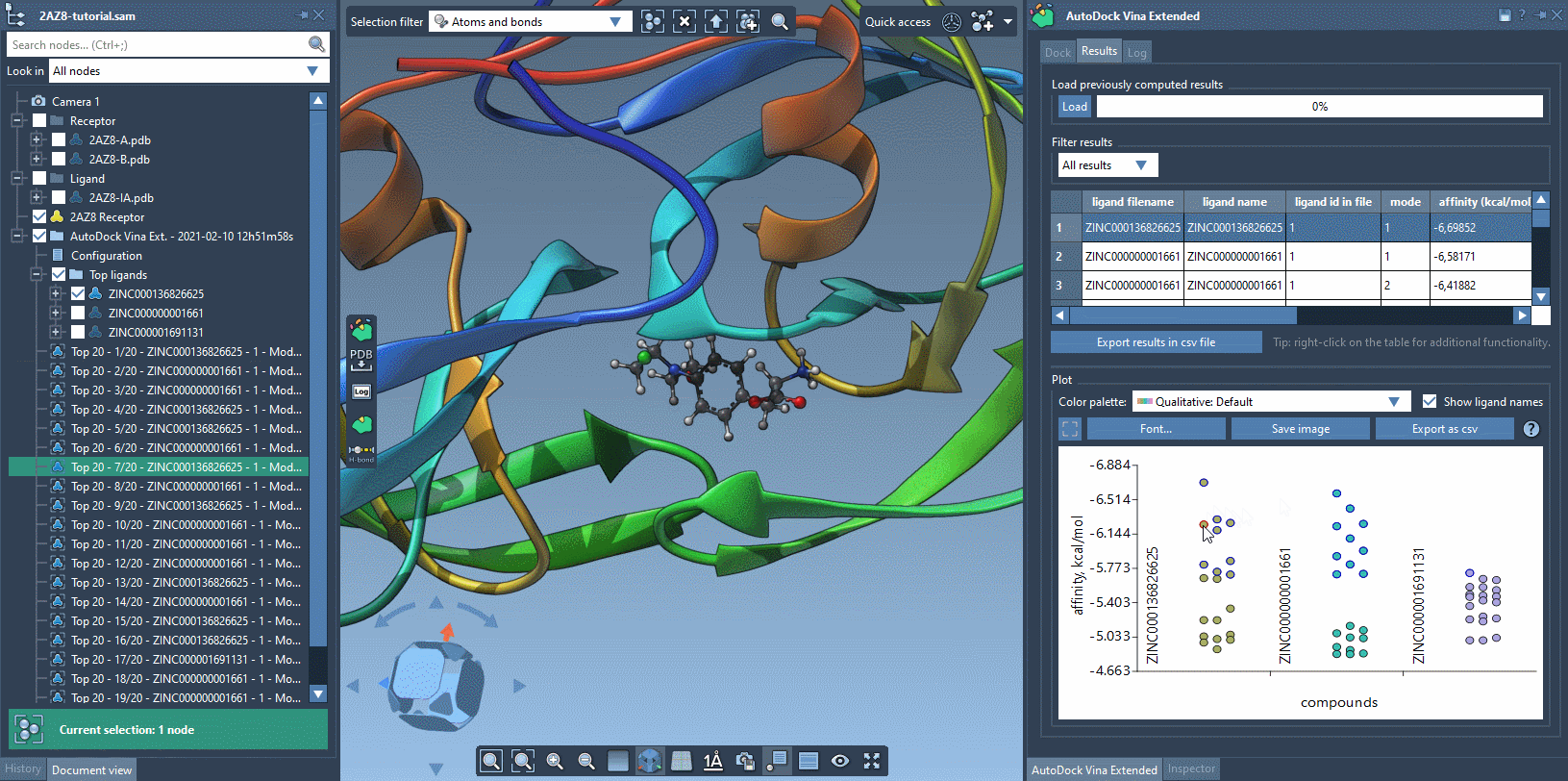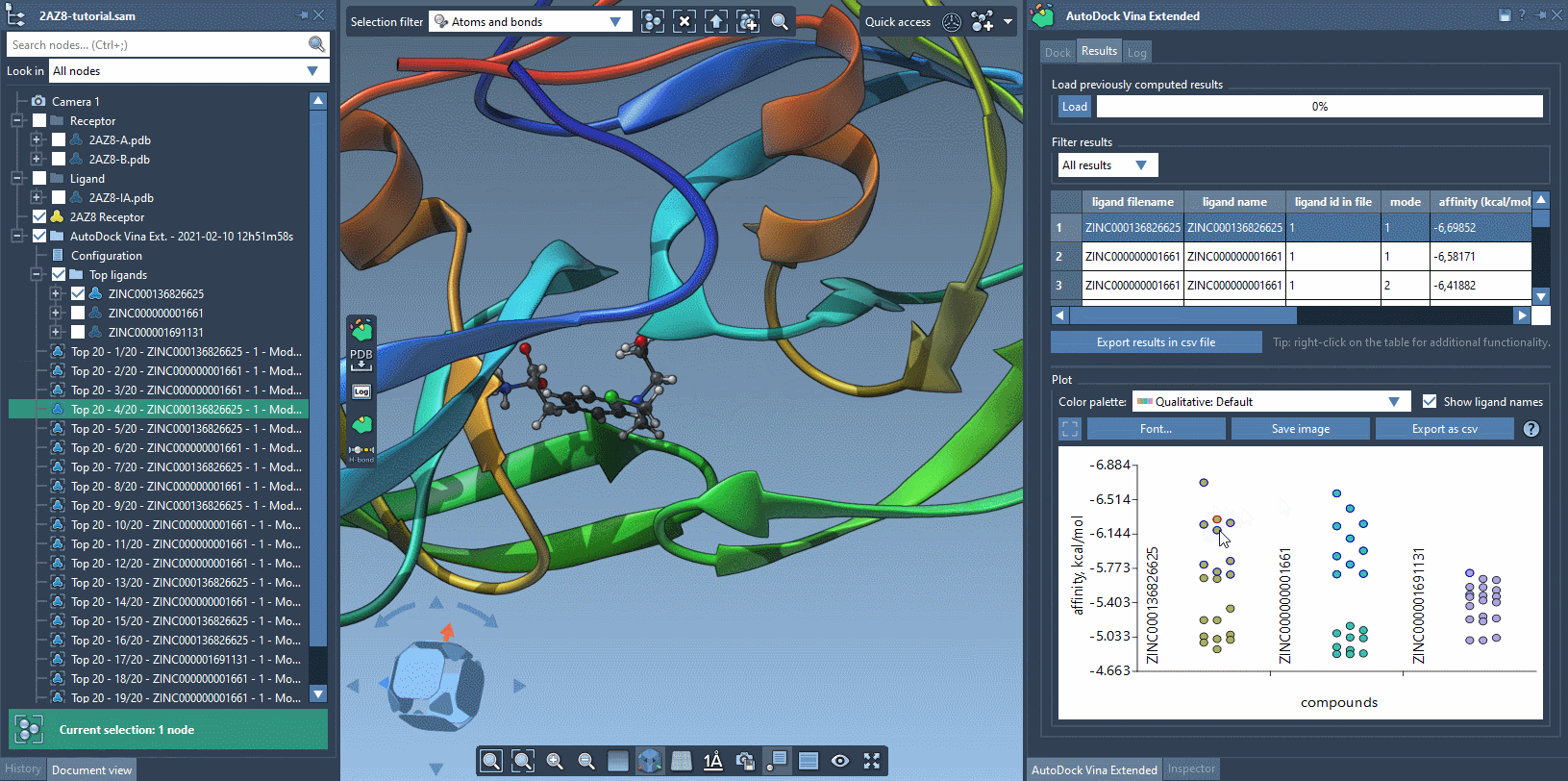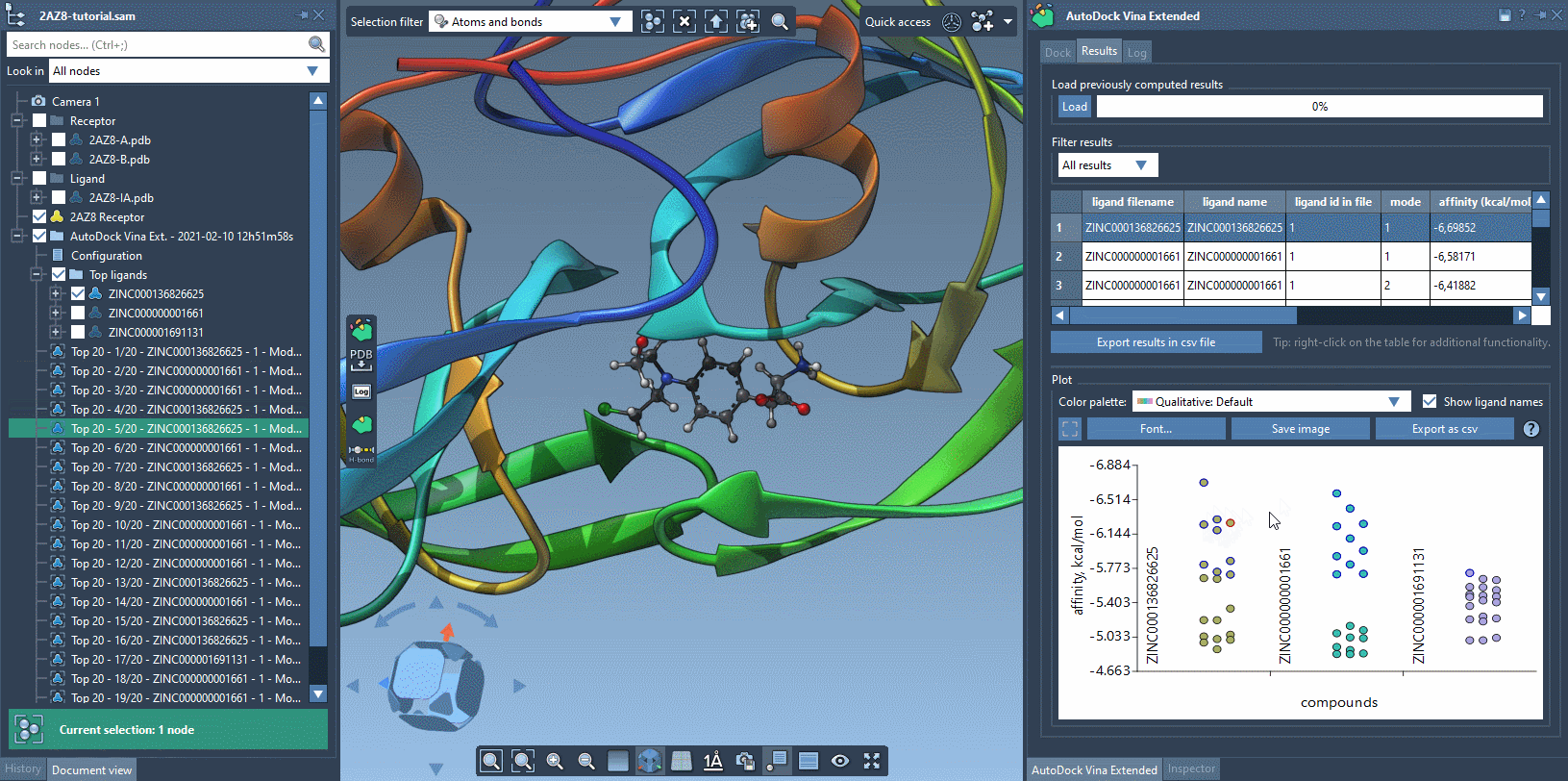
You can use the filter to modify what is shown both in the results table and in the plot.
Loading previously computed results#
You can load the previously computed results obtained with AutoDock Vina Extended using the Load functionality in the Results tab. Please note that you shouldn't modify the hierarchy of the project directory or remove files if you want to be able to load them back into AutoDock Vina Extended. There are several options for loading the results:
- Only the table of results (fast) - loads only the table of results without loading molecules and conformations. Ligands and their conformations can be loaded later from the results table.
- Top ligands - loads results (molecules and conformations) only for the specified number of top ligands (by score). The other ligands and their conformations can be loaded later from the results table.
- Select results to load - loads results (molecules and conformations) only for the selected ligands - a pop-up dialog will appear from which you can select the results to load. The other ligands and their conformations can be loaded later from the results table.
- All results (slow) - loads all the modes for all the ligands. Please note that it might take time to load docking results for a big ligand library.
Let's now try to load the results that we just computed. Open a new document (Ctrl/Cmd + 3 + N). Click on the Load button and navigate to the directory with the results:
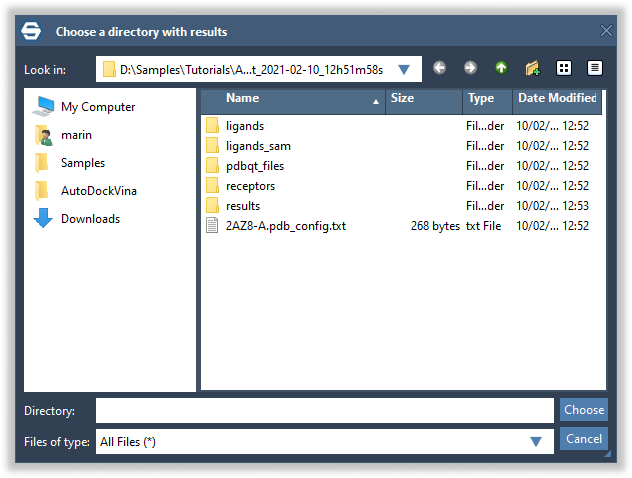
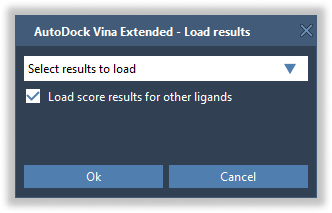
In the next pop-up dialog, select Select results to load from the list and check the Load score results for other ligands option to load score results for the non-selected ligands in the results table as well.
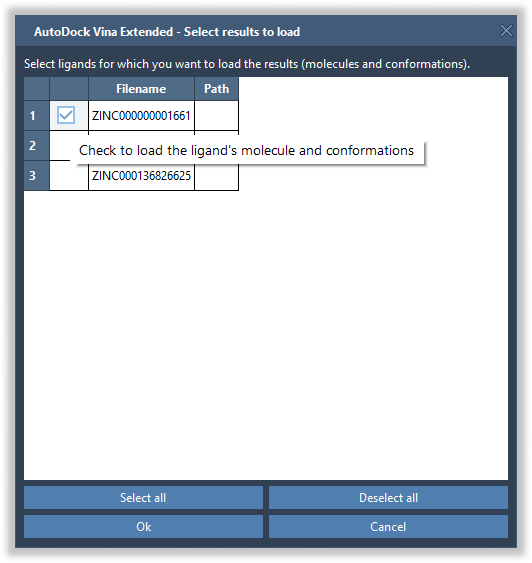
Another dialog should appear with a list of ligands found in the results directory. You can deselect all of them by clicking on the Deselect all button and then check the ones which you want to load. Please note that loading results for many ligands might take some time. You can later load the other ligands and their conformations from the results table (see below).
A new folder with the results should appear in the document, containing two subfolders:
Receptorswith a receptor and groups with flexible residues and side chains if they were specified.Docked ligandswith subfolders for each loaded ligand, each containing a ligand and its conformations.
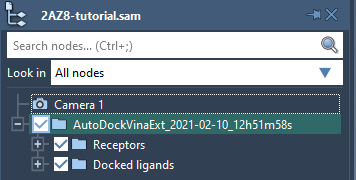
If you do not see the loaded receptor or ligands in the document, please make sure that their structural models are checked in the Document view as shown in the image below.
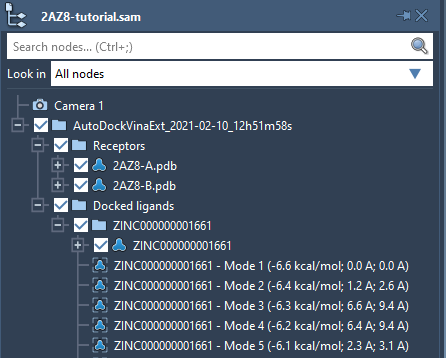
You can load results for other ligands in the results table by right-clicking on a ligand for which you want to load the results and choosing Load ligand results from the context menu. This will load the ligand and its conformations in a subfolder with the ligand's name in the Docked ligands folder.
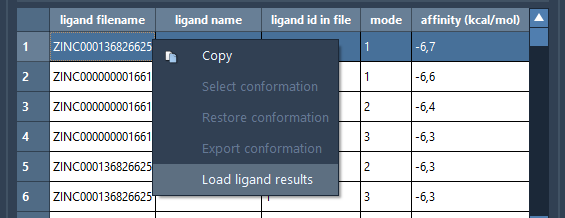
Performing further analysis#
SAMSON provides different tools to perform analysis of the results. You can add various visual models, measure distances, compute some parameters, and check for ligand-receptor interactions. Below you will find some examples.
Visualizing#
Let's first add some visual models for the system.
You can apply a visual preset in one click using Visualization > Visual preset - select Protein-ligand visual preset.
Or you can apply visual models yourself - follow the instructions below.
Tip
You can learn more about visualization in SAMSON thanks to the interactive tutorials (Help > Tutorials) or from the User guide: Visualizing.
First, select the receptor and add a secondary structure visual model to it using Visualization > Visual model > Ribbons. This should add a new secondary structure visual model to the document.
A hint: if you want to hide a visual model, simply uncheck it in the Document view; if you want to remove it then right-click on it and choose Erase from the context menu.
Let's now add labels and a licorice visual model for residues surrounding the ligand. First, we need to select residues surrounding the ligand. For that, select the ligand and click on Select > Biology > Binding sites and set the parameters as in the image below, and click OK. This will select the residues surrounding the currently selected ligand.
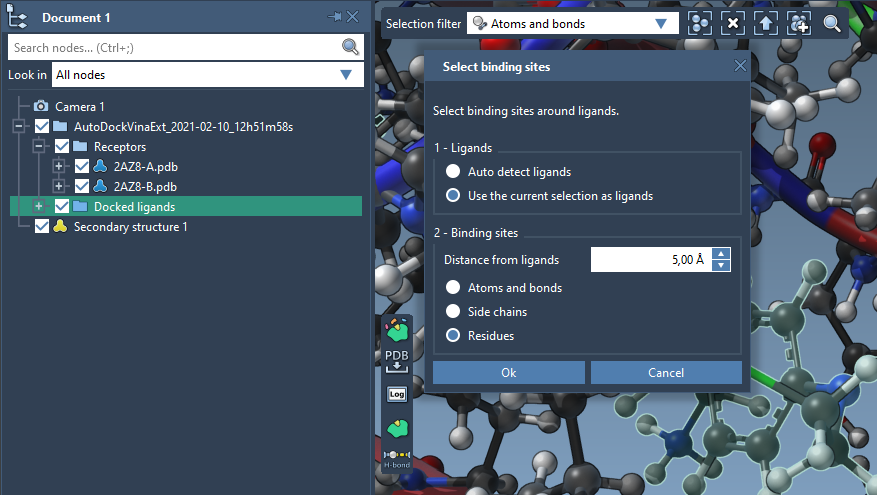
You can save this selection in a group by clicking on the Current selection button in the Document view and clicking on Create group in the context menu or on Select > Group.
While having these residues selected, click in the context toolbar on ![]() to add labels and choose Add label to... > Residues. This will add in the document a new folder called
to add labels and choose Add label to... > Residues. This will add in the document a new folder called Residue labels. You can easily hide labels by either unchecking them in the document or by deleting them.
Tip
See User Guide: Labeling.
Let's now add a licorice visual for these residues. While having these residues selected, go to the Visualization > Visual model > Licorice - this will add a licorice visual model for the selected residues.
If you want, you can colorize carbons in the receptor based on e.g. the residue sequence number. For that, select the receptor (Select > Biology > Receptor) and then go to Select > Atoms > Carbons.
This will select all carbons in the current selection. Let's now colorize them by the residue sequence number such that their color would correspond to the default style of secondary structure colorization. For that, choose from the Visualization > Color > Per attribute menu the Residue index option. This should colorize both the carbons in the receptor's structural model and in the added licorice visual model since the licorice visual model doesn't have a specific color scheme applied to it.
Now let's hide the receptor's structural model by unchecking it in the document. In the end, you should see something like in the image below:
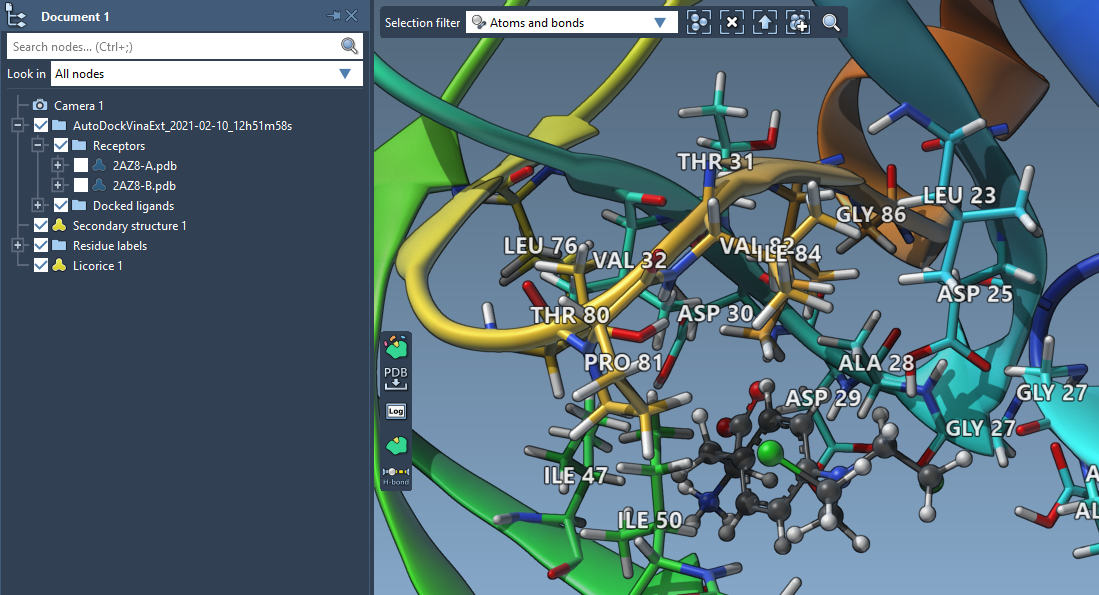
Please note that you can modify the appearance of visual models using the Inspector.
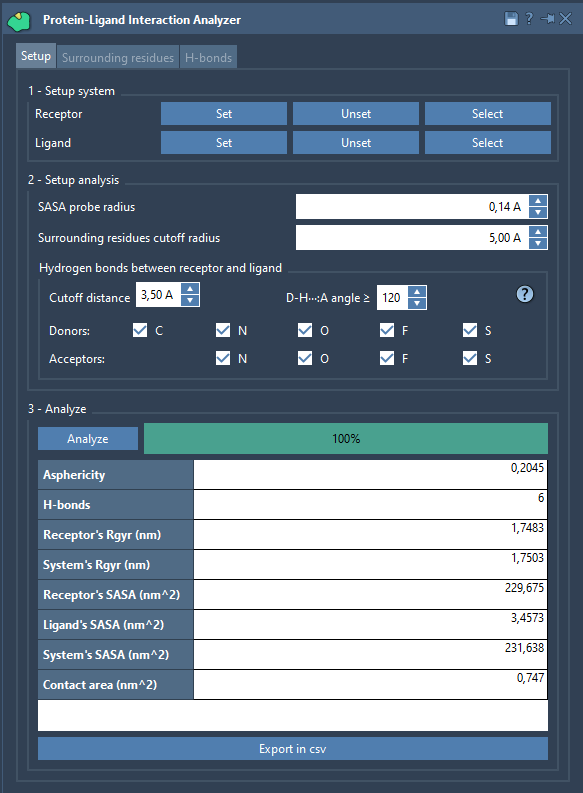
Protein-Ligand Interaction Analyzer#
The Protein-Ligand Interaction Analyzer SAMSON Extension allows for computing the contact area, H-bonds between a ligand and a receptor, ligand surrounding residues, and some other parameters. Check out its other tabs for more functionality.
Hydrogen bonds#
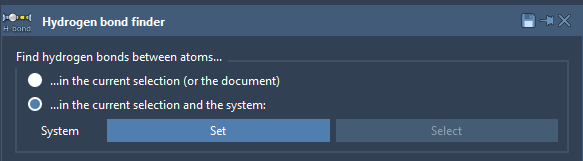
The Hydrogen Bond Finder SAMSON Extension allows one to find and visualize hydrogen bonds (H-bonds) inside a molecule or between molecules, e.g., between a ligand and a receptor. To find H-bonds between a ligand and a receptor, select the second option ("... in the current selection and the system"), then select the receptor and click on the Set button, modify parameters if necessary (you can later modify them in the created H-bond visual model using the Inspector), then select a ligand and click on the Add hydrogen bond visual model button.
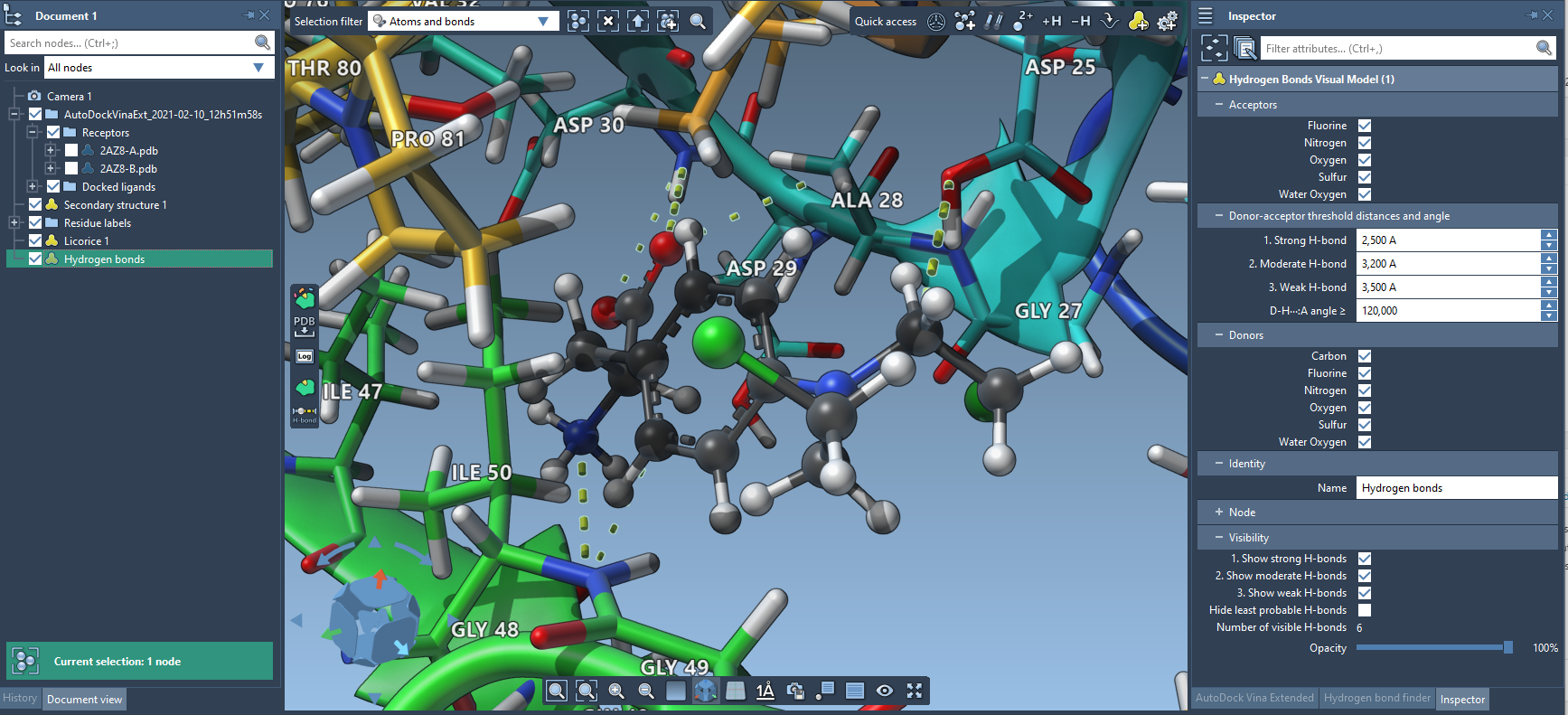
This will add a new H-bond visual model to the document. If you want to modify the H-bond detection parameters, right-click on the newly created H-bond visual model and choose Inspect from the context menu - this will open the Inspector in which you can modify parameters.
That's all, thank you for completing this tutorial on the AutoDock Vina Extended SAMSON Extension.
Related tutorials#
- Protein preparation and validation for preparing protein structures before docking.
- Generate analogue series with positional analogue scanning for creating ligand analogs before docking.
- Strain Explorer for comparing ligand strain after docking or conformer generation.
Next step#
Use the docking poses and scores to choose candidates for interaction analysis, ligand refinement, or molecular dynamics with GROMACS Wizard protein-ligand systems.
Need Help?#
Have questions or feedback? Feel free to reach out via the Forum, via e-mail, via the Feedback button in SAMSON, or by directly discussing with us.