Universal Force Field#
In this tutorial, you will learn how to set up and use the Universal Force Field (UFF) interaction model in SAMSON.
This interaction model implements the Universal Force Field (UFF) proposed by Rappe et al. 1. It also includes an automatic scheme to perceive the molecular system in order to compute the bonds, bond orders, and atom types, so that UFF can be applied directly to a given molecular system. For more details regarding this UFF implementation and the approach used to automatically perceive the molecular structure, please refer to publication 2.
What you will learn#
In this tutorial, you will learn how to set up, run, and customize the Universal Force Field interaction model in SAMSON.
Before you start#
- Open a molecular system that you want to simulate or edit interactively.
- If you are new to SAMSON simulation tools, keep the workflow simple the first time and use the default settings.
- Use this page as a practical getting-started guide; the references remain the best source for the deeper theory.
Setting UFF#
- Open a document with a molecular system you want to simulate with UFF.
- Add a simulator via Edit > Simulate > Add simulator (or use the shortcut Ctrl+Shift+M / Cmd+Shift+M).
- Select Universal Force Field in the list of interaction models.
- Choose the state updater you want to rely on for the simulation (e.g., FIRE).
- Press the OK button.
The UFF setup window now appears.
- Check Use existing bonds if you want to rely on the existing bonds of the model. Otherwise, UFF will recompute the bonds from the atom types and positions.
- Press the OK button.
The module now initializes the system for UFF by performing the following operations:
- Compute the covalent bonds if Use existing bonds was not checked earlier.
- Compute the bond orders.
- Compute the atom types.
In case of detected inconsistencies, a warning message describing these inconsistencies may appear.
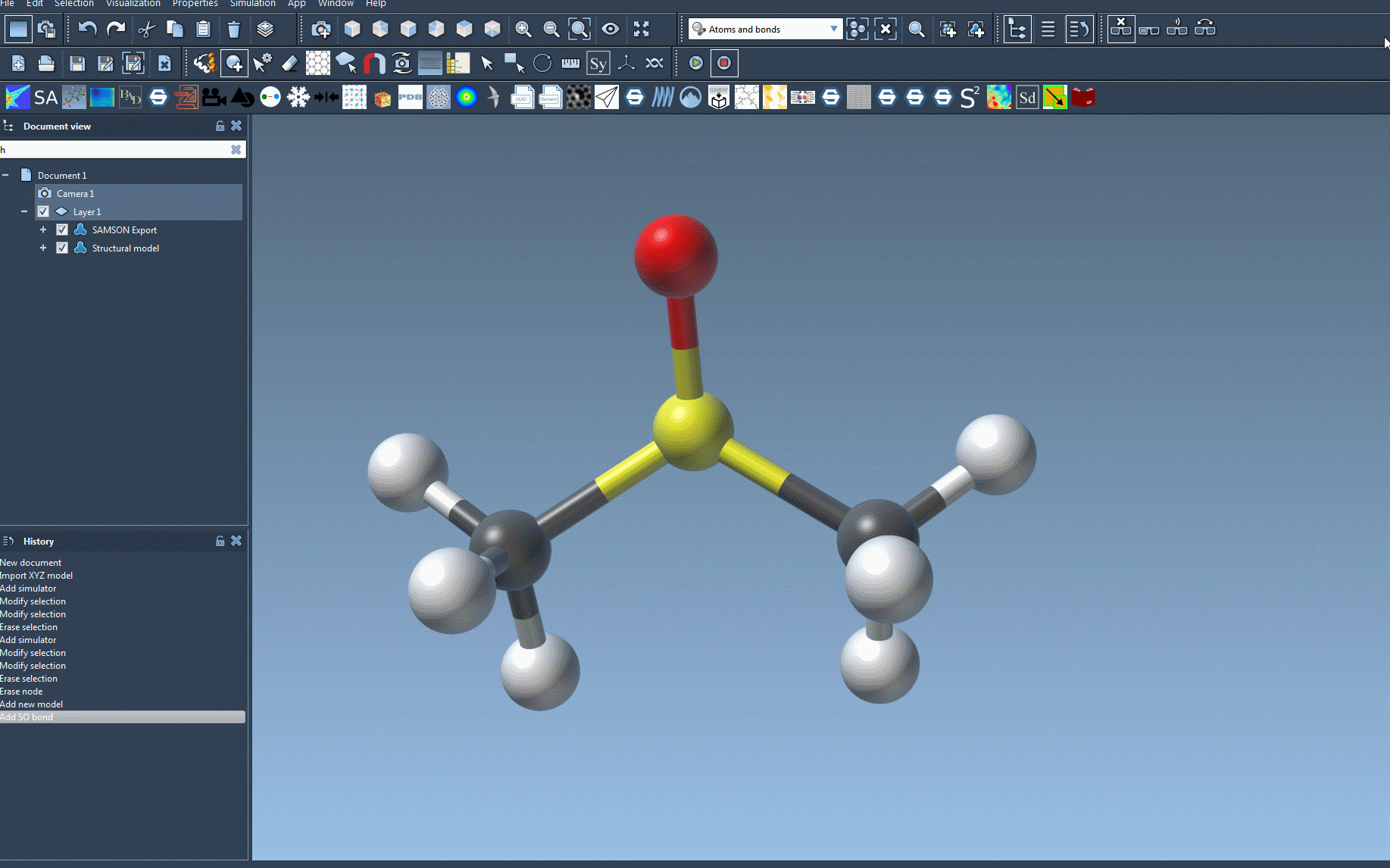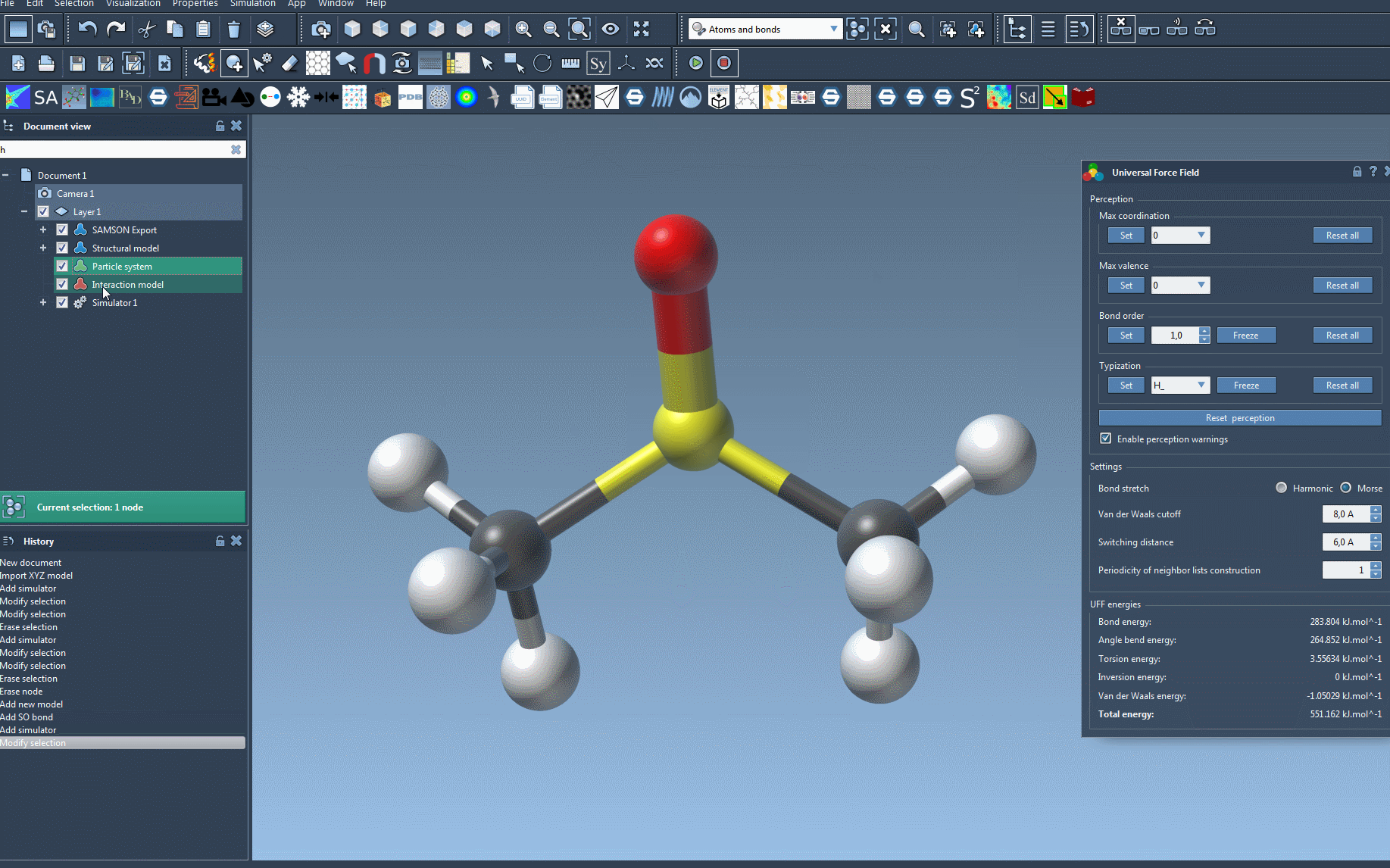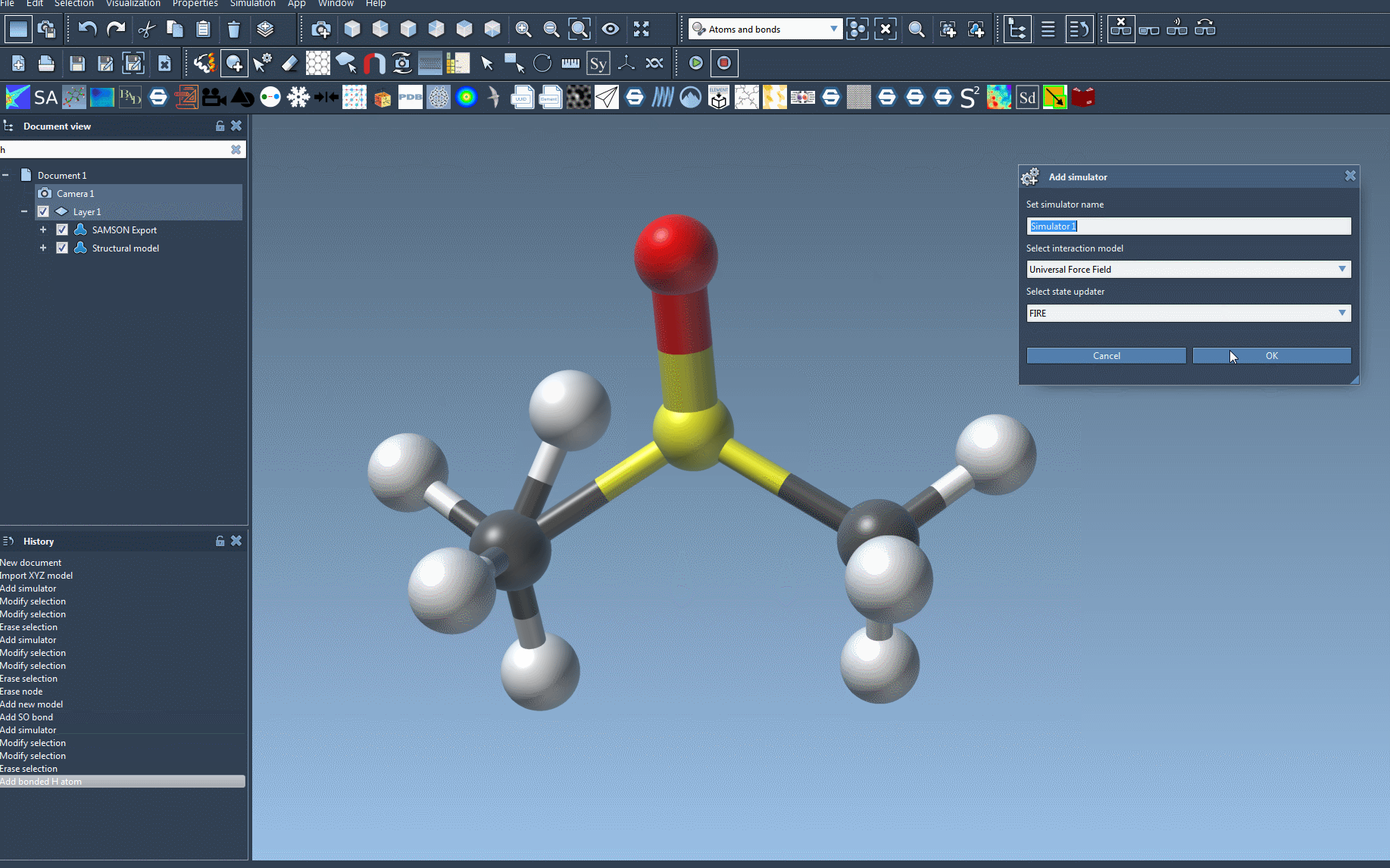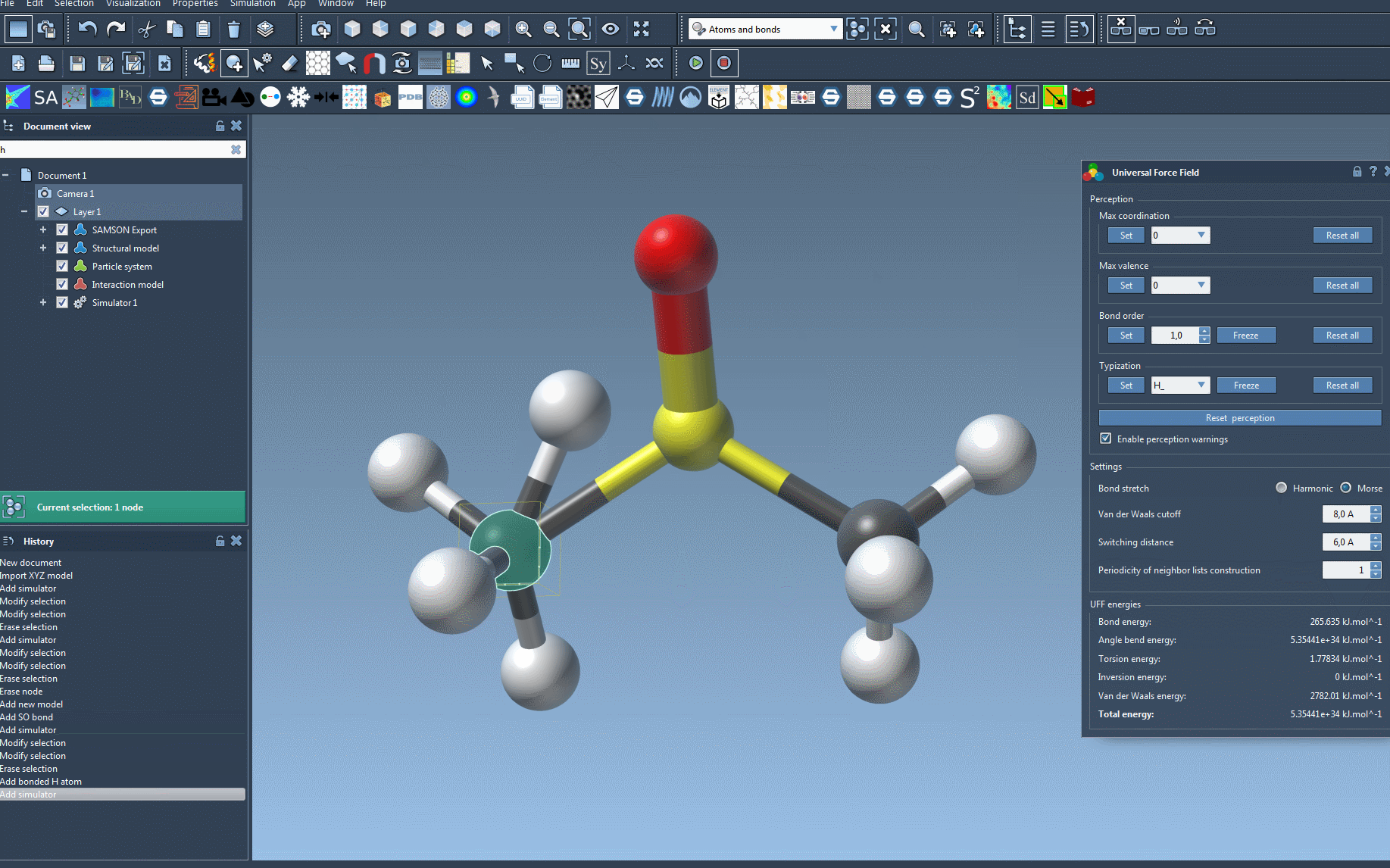
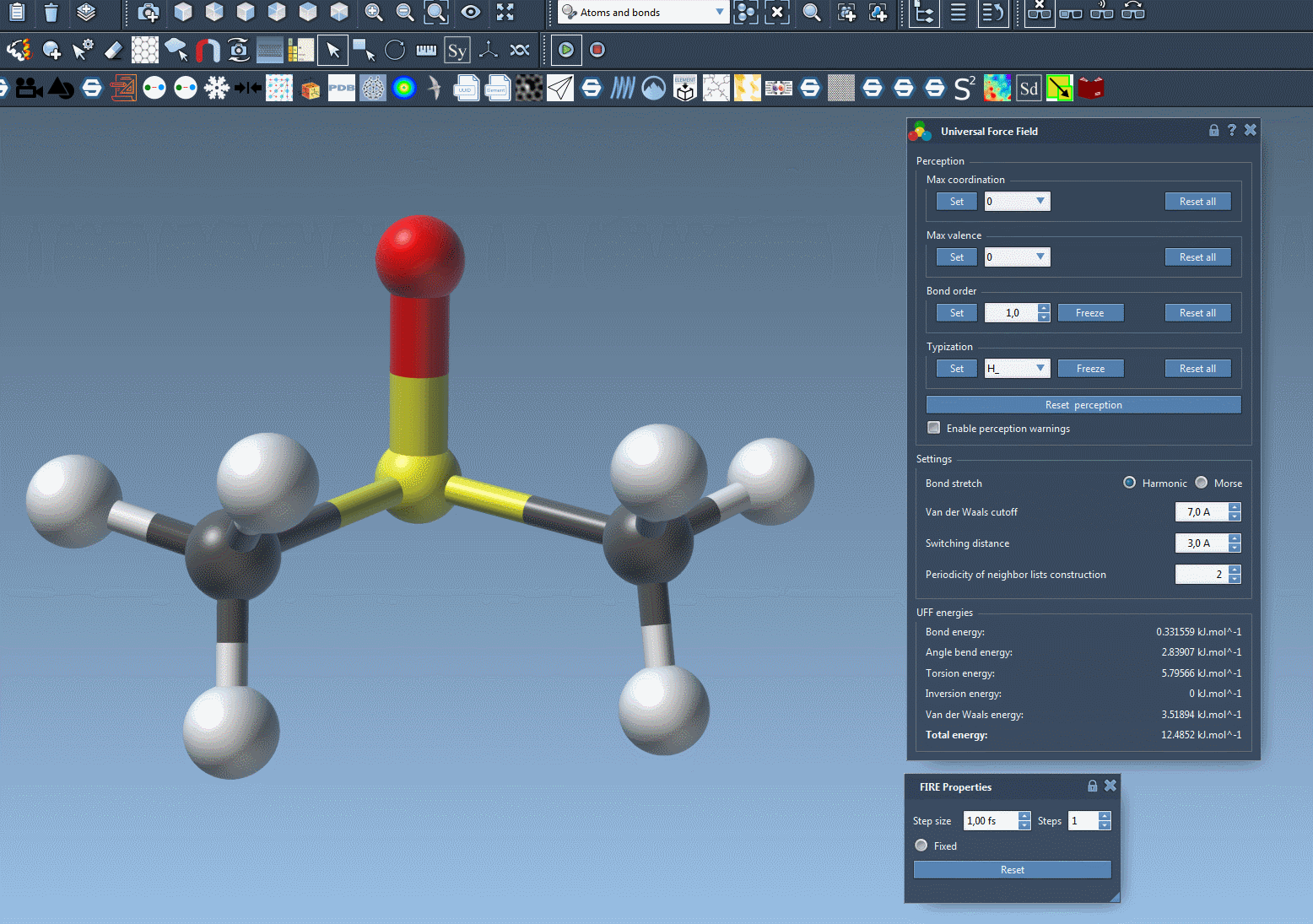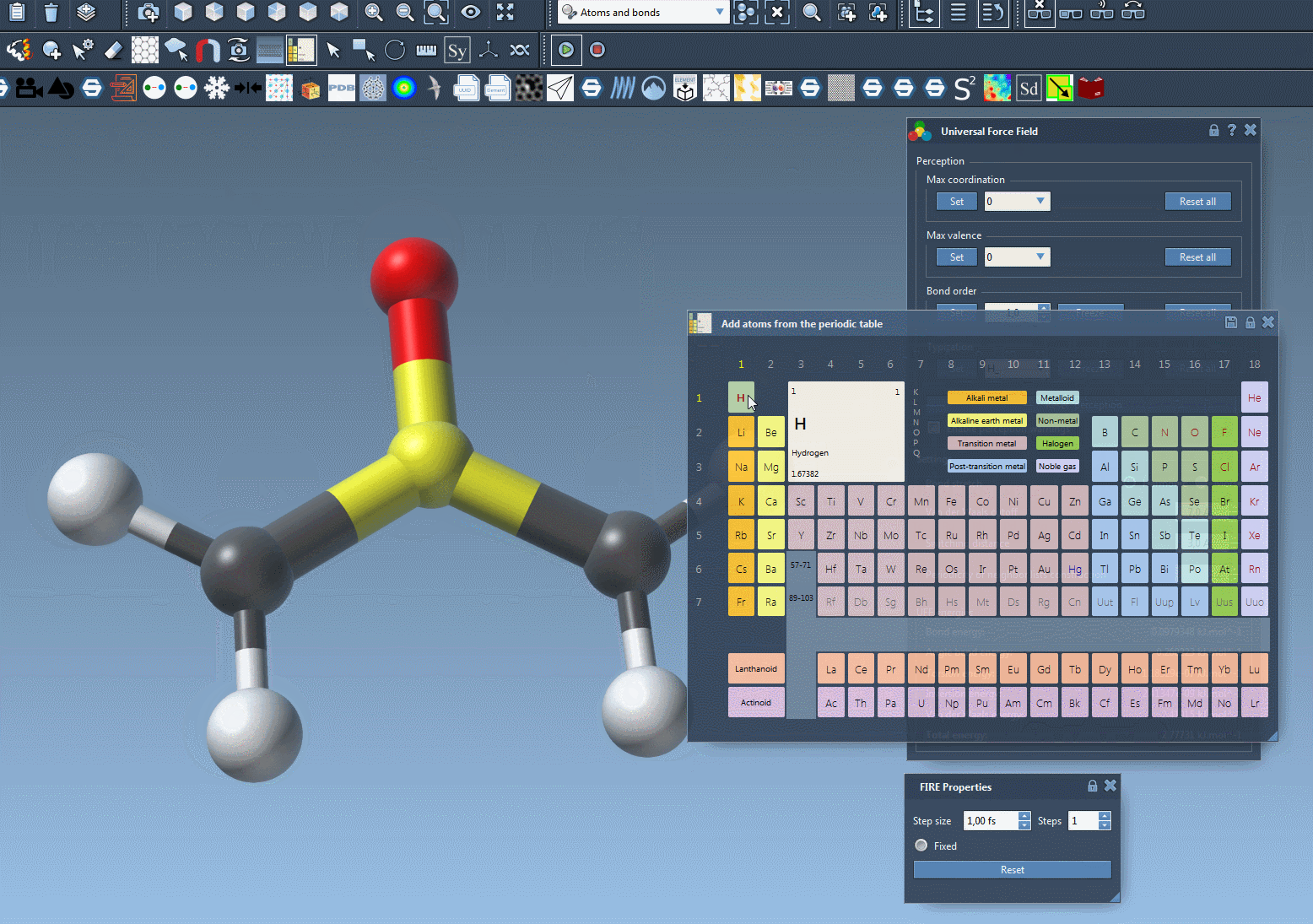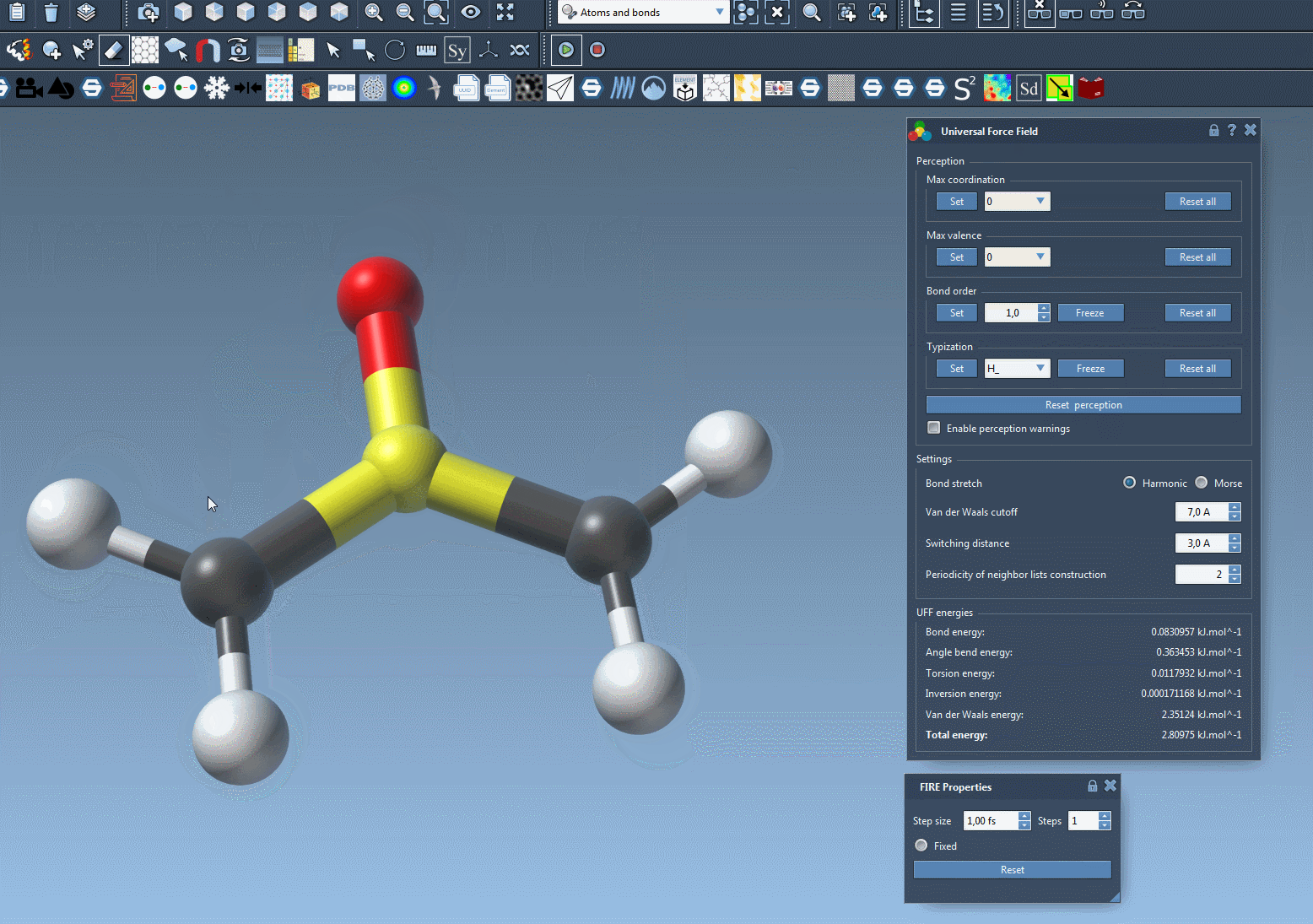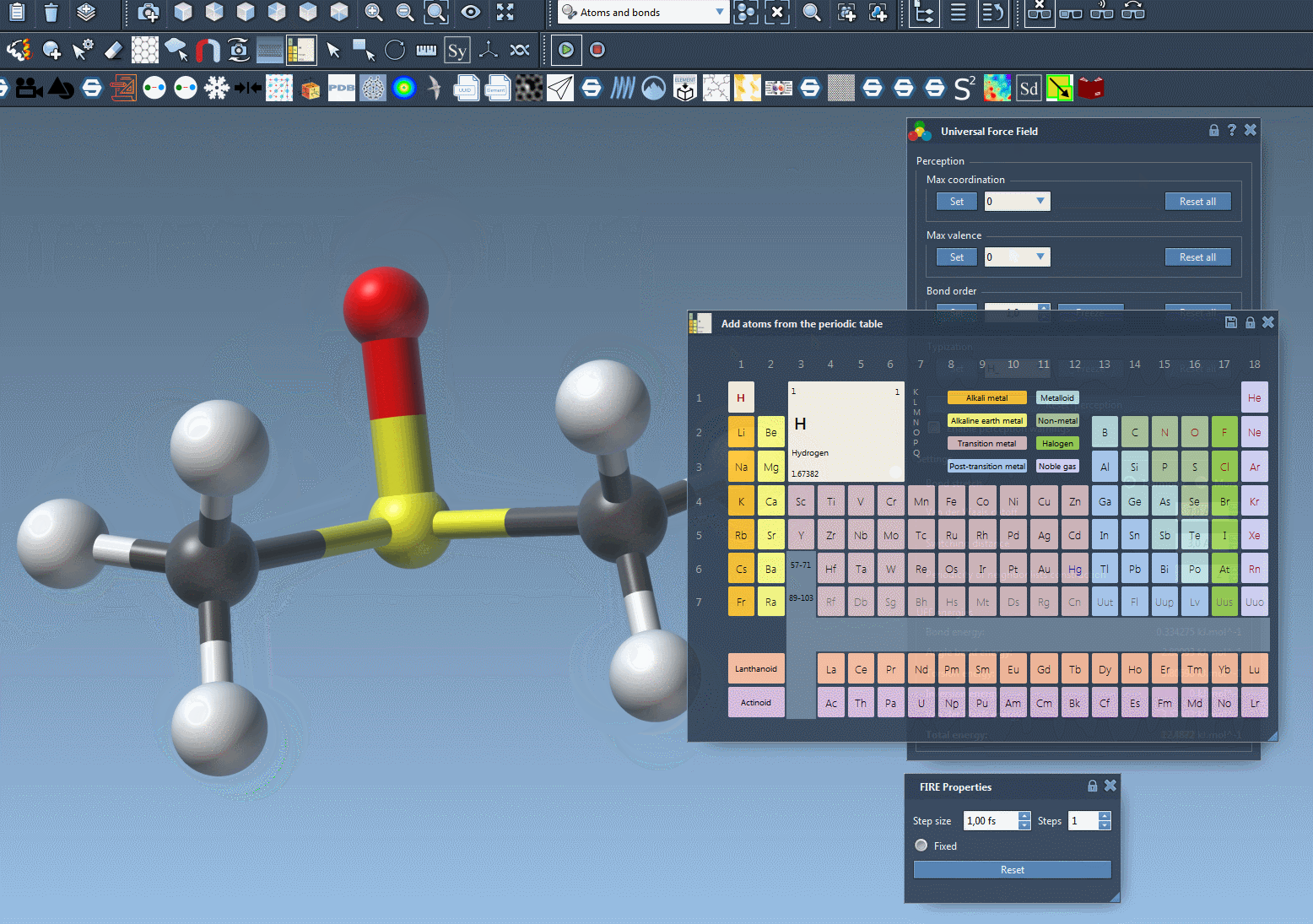
At this stage, you should now see the parameter window of UFF.
Running the UFF simulation#
You can now run the UFF simulation:
- Press the Edit > Simulate > Start button.
A UFF simulation is now running. At the bottom of the UFF window, you can see the energy of each UFF term as well as the total UFF energy. You can interactively move atoms and see how the UFF model reacts by updating the position of the other atoms as a consequence.
The UFF interface allows you to modify the following UFF parameters:
- the type of bond stretch interaction: it is possible to switch between Harmonic and Morse for this interaction by selecting the corresponding radio button (see the detail of the difference between these modes in 1).
- the van der Waals cutoff distance that limits the distance for which vdW interactions are considered.
- the switching distance from which the vdW term is weighted so that it smoothly reaches zero at the cutoff distance.
- the periodicity of neighbor lists construction, that may be modified to recompute the neighbor lists used in vdW more or less often (by default it is recomputed at each time step).
Customizing the typization#
Several options are offered to customize the typization of UFF in SAMSON. Note that such customization is recommended for advanced users only since it can easily lead to incorrect structures in case of misuse. Below we describe the options proposed to customize the UFF typization.
- Some default maximum coordination (i.e., the number of neighbors) and maximum Valence (i.e., the number of neighbors weighted by their bond order) are set for the atoms (see 2 for more details). These default values can be modified for each atom by selecting the new value in the corresponding combo box and pushing the Set button. This new value is then taken into account in the new round of automatic atom perception. Note that these values are effective only if the new maximum coordination of valence is lower than the default one.
- The bond order of the bonds can be forced. For this, first, select the bond (or set of bonds) you are interested in, then select a bond order value (between 0.1 and 3.9) in the UFF parameter window, finally, push the Set button in the bond order group. You can also force a bond order to not change after a new round of perception. For this, first, select the bond(s) and then press the Freeze button of the Bond order group in the UFF parameter window. Note that non-integer bond orders are possible.
- Similarly, the UFF types of atoms can be forced. For this, first, select the atom (or set of atoms) you are interested in, then select the UFF type you want to assign to the atom(s). Finally, push the Set button to force the type. You can also force a given type to not change after a new round of perception. For this, select the atom(s) and press the Freeze button of the Typization group in the UFF parameter window.
You can reset all of the previously mentioned customizations by pressing the corresponding Reset all buttons. Finally, you can recompute a new perception from the current customized settings and current atom positions (since bond creations depend on atomic distances) by pushing the Reset perception button.
Deleting and adding atoms#
Atoms can be deleted from the model and added to the model "on the fly" i.e., the typization is then recomputed automatically accordingly.
- Deleting an atom: select the eraser editor (rubber icon), then, select the atoms you want to delete.
- Adding an atom. Add atoms connected to this system by creating them as bonded to an existing atom of the model. e.g.: select an atom element in the periodic table, editor; click on a simulated atom, move the mouse, you should see an atom bonded to the initial atom appearing in transparency, then release the button of the mouse after moving the mouse. The atom should be part of the simulated model.
Tip
You can learn more about how to build systems from User Guide: Building molecules or the Building with atoms interactive tutorial in SAMSON (Help > Tutorials).
That's all for this tutorial. For more details about the UFF implementation, please see reference 2.
Related tutorials#
- Interactive Modeling Universal Force Field for interactive atom typing during model editing.
- Fast geometry optimization with FIRE minimizer for geometry optimization with a state updater.
- UMA Force Field for a machine-learning based alternative to classical force fields.
Next step#
Continue with Interactive Modeling Universal Force Field if you want a more interactive UFF-based workflow.
Need Help?#
Have questions or feedback? Feel free to reach out via the Forum, via e-mail, via the Feedback button in SAMSON, or by directly discussing with us.
References#
-
A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. Goddard III and W. M. Skiff. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. Journal of the American Chemical Society 114.25 (1992): 10024-10035. ↩↩
-
S. Artemova, L. Jaillet and S. Redon. Automatic molecular structure perception for the universal force field. J. Comput. Chem. 2016, 37, 1191-1205. ↩↩↩