Generate crystal models#
Use the Crystal Creator Extension in SAMSON to import CIF files, inspect crystal properties, cut crystal slabs, and build your own periodic models from unit-cell data.
What you will learn#
In this tutorial, you will learn how to import CIF files, inspect crystals, and build crystal models in SAMSON with Crystal Creator.
Before you start#
- Add Crystal Creator from SAMSON Connect - Marketplace if it is not already installed.
- If you want to follow the import workflow, download a CIF file from one of the sources listed below.
- Use this page both as a first tutorial and as a reference when you need to build a custom crystal later on.
Read a crystal structure#
To have your first crystal, you can import one. With the Crystal Creator App, you can load CIF (Crystallographic Information File) files. First, go fetch some nice crystals on those two websites:
- The American Mineralogist Crystal Structure Database: https://rruff.geo.arizona.edu/AMS/amcsd.php
- The RRUFF Project Database: https://rruff.info/
I personally recommend testing Macdonaldite, Quartz, and Zorite, but if you have a favorite crystal and you find it, test it.
Open your CIF file in SAMSON. A window lets you choose the number of crystal unit cells, if you prefer to import the mere data without the symmetries, and if you want to see the mesh. Click on Open and the structural model of the crystal and its property model appear in the data graph (see User guide: Loading molecules if you have problems opening files in SAMSON).
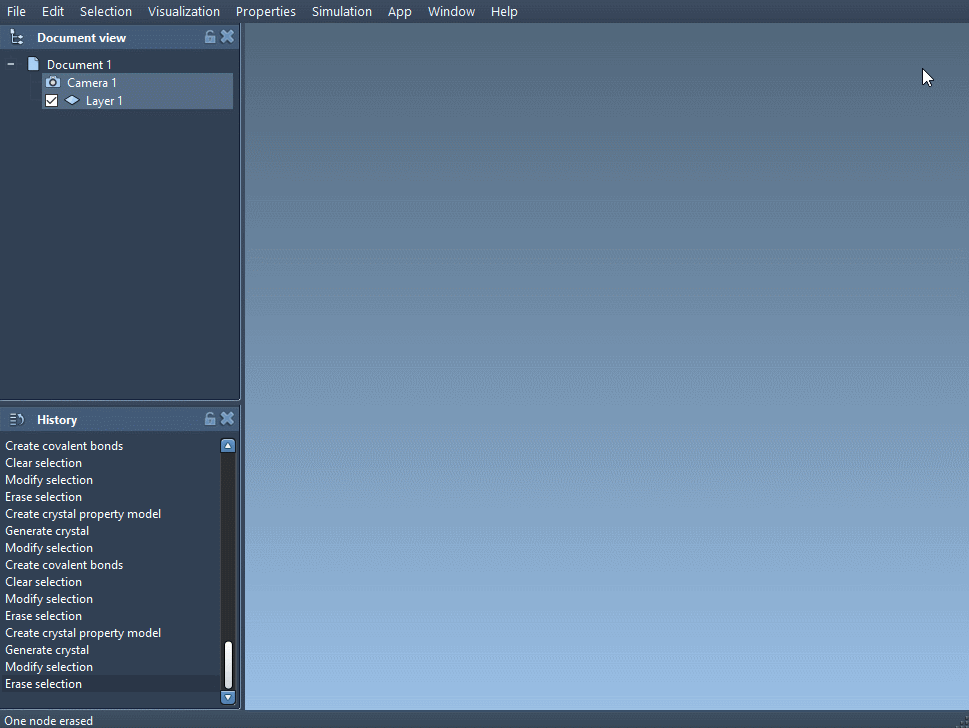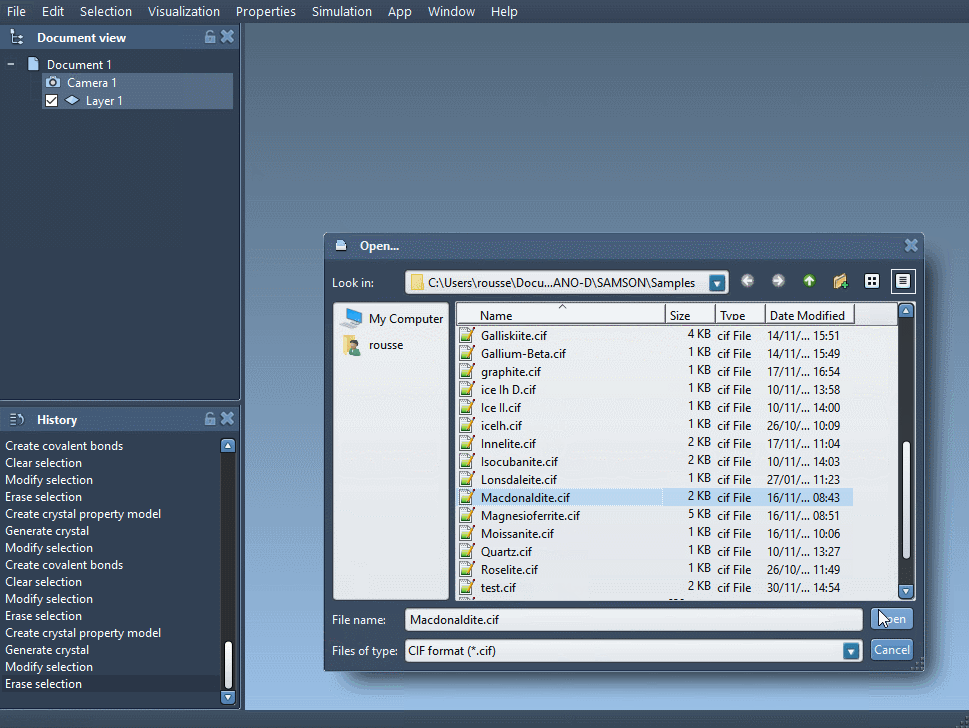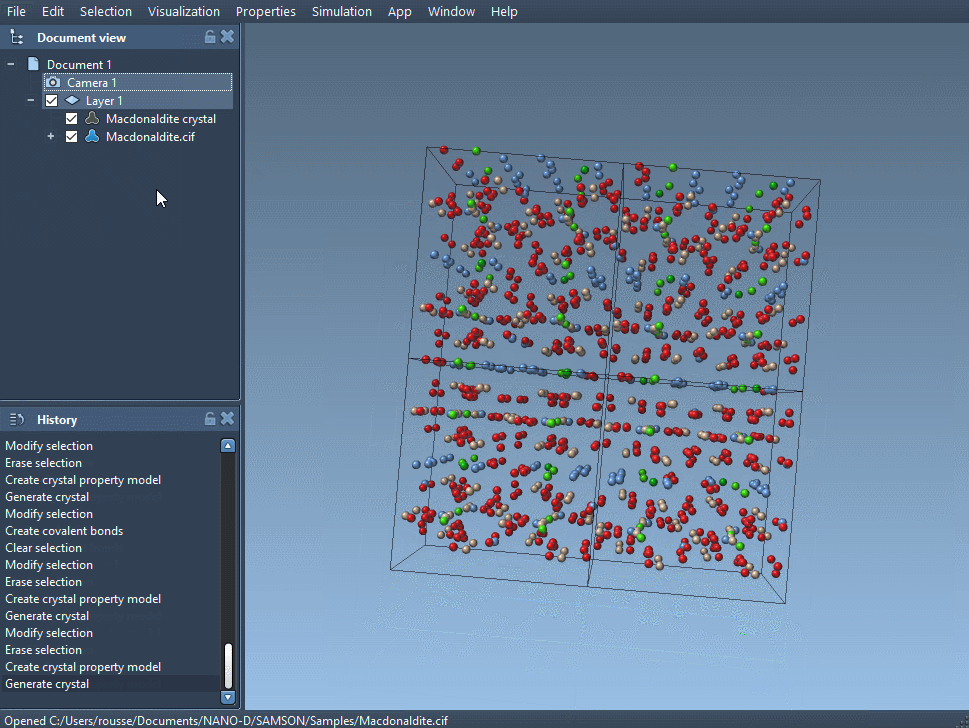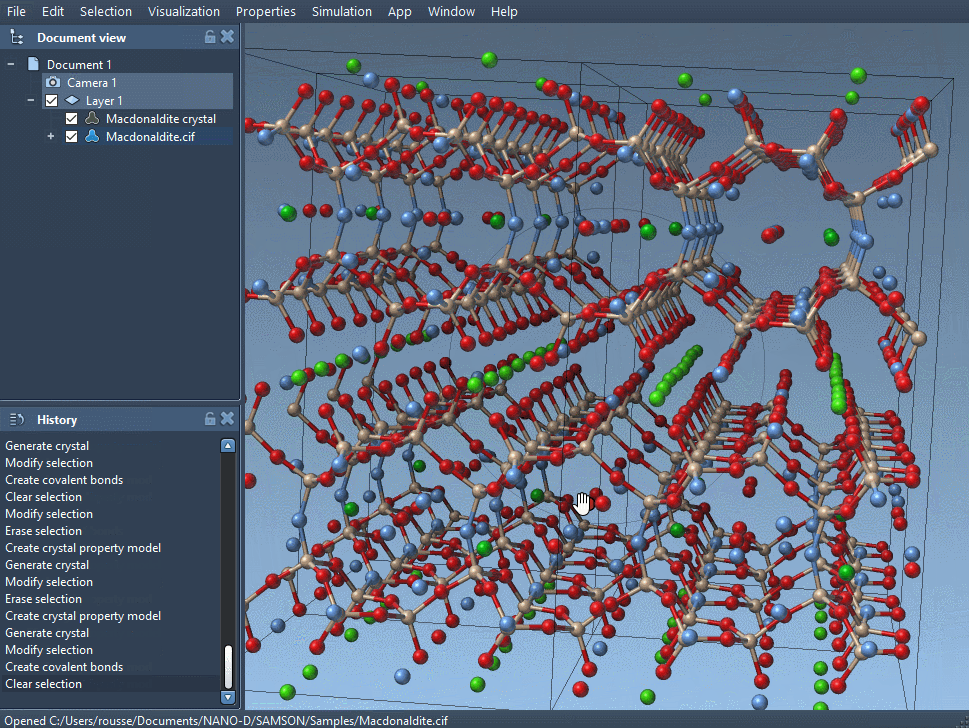
Depending on the crystal you have just read, the unit cells of your crystal can be different. Indeed, interesting properties of a crystal appear with randomly distributed defects or substitutions; those are described in the CIF file and are used by the app to create a crystal with proper defects and substitutions.
Manipulate a crystal#
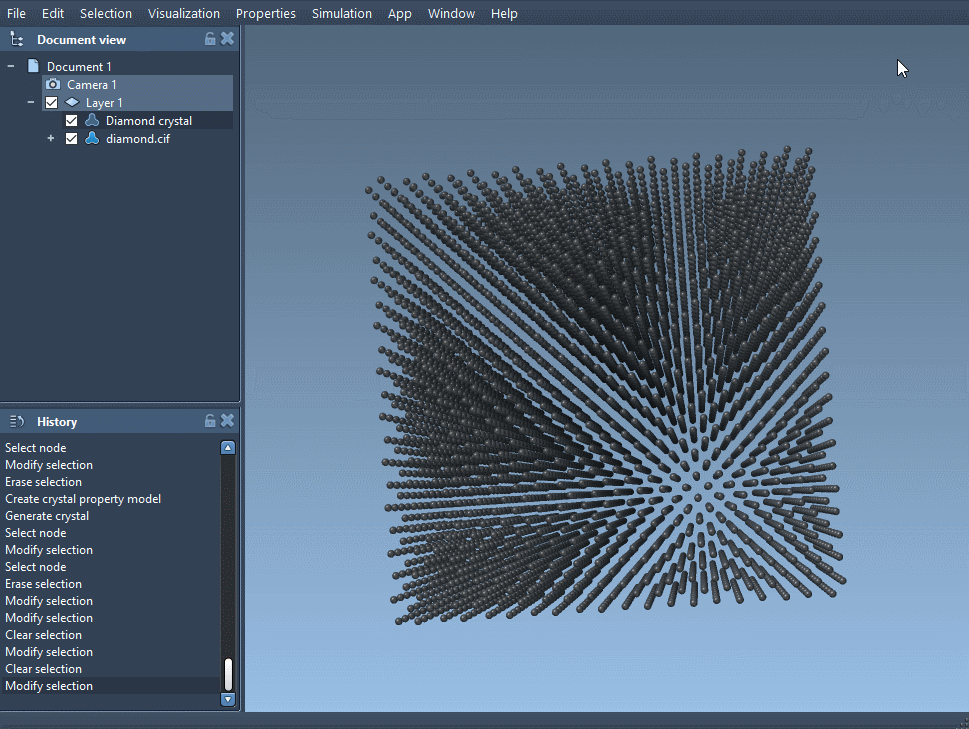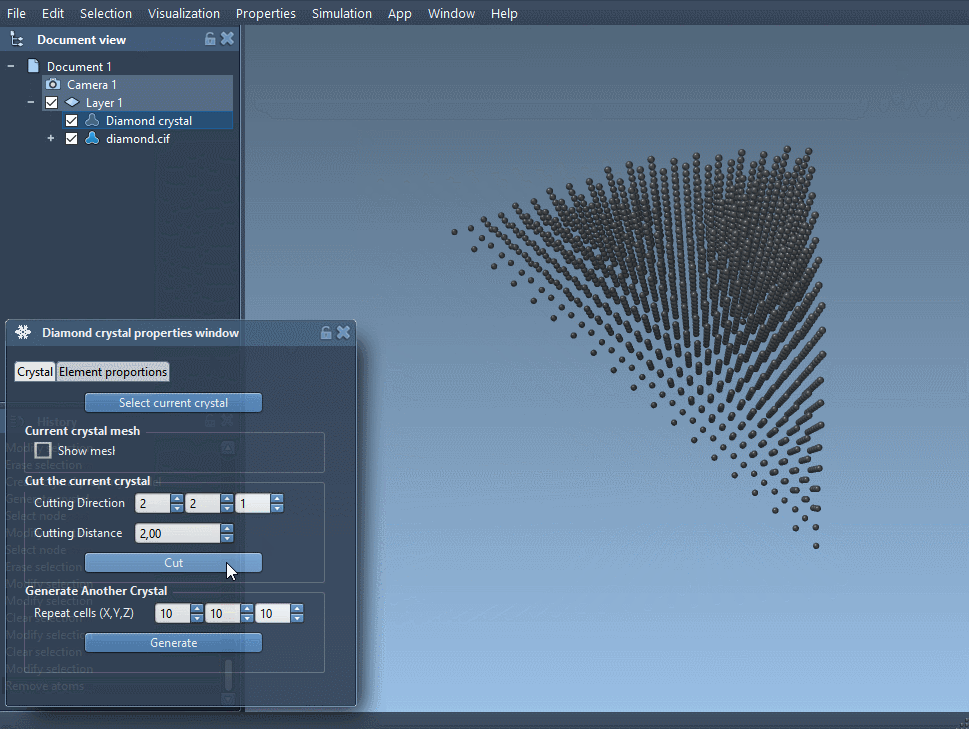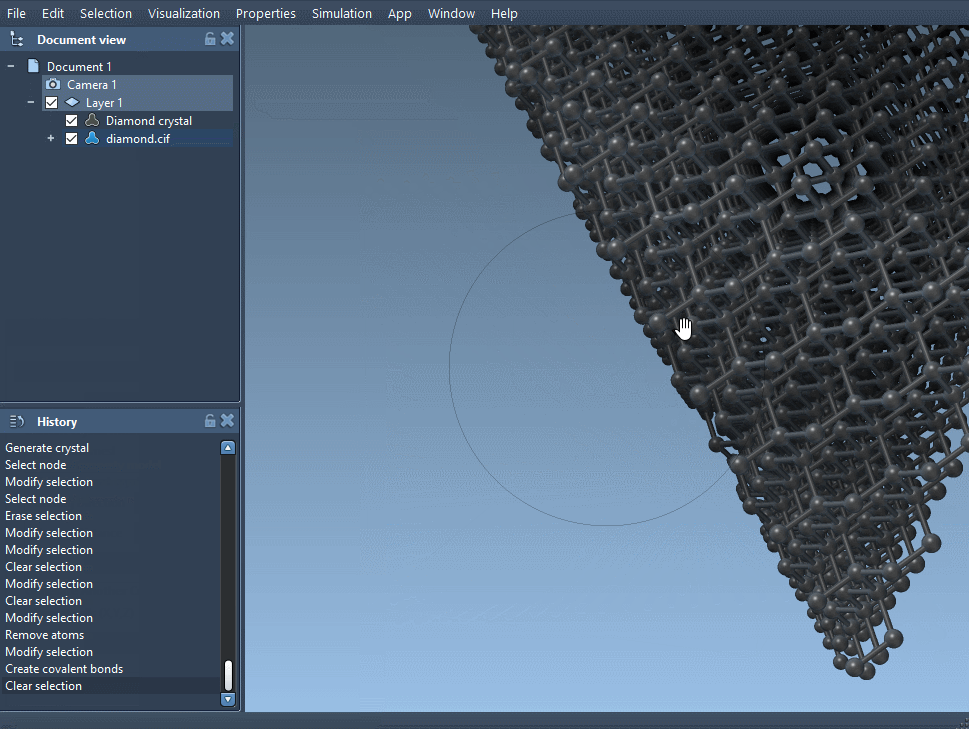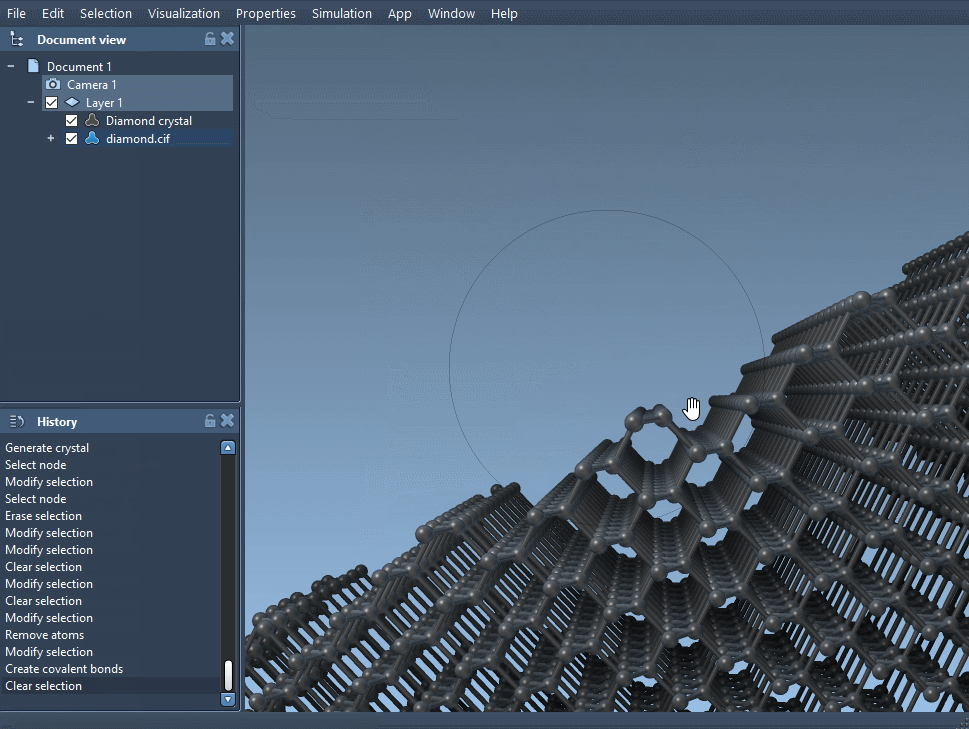
Right-click on the property model and select properties.
The first tab of this property window has 4 tools:
- A button to find back your crystal.
- A checkbox to see its mesh.
- A toolbox to cut your crystal. The first three boxes describe the cutting direction (the Miller indices) and the fourth is the distance from position 0 where the cut is done.
- A toolbox to generate another crystal.
The second tab is a tool to check if the generated crystal contains the proper amount of defects and substitutions. By clicking on check atoms ratio, you can see, for each atom site, whether its ratio of presence/absence is respected.
Download and open a diamond crystal file and cut it in the direction [111] to see its compact hexagonal structure.
Create your own crystal#
Open the Crystal Creator App via Home > Apps > Materials > Crystal Creator or search for it in the top search box (Shift+E). In this app, you can:
- Choose the position 0 of your crystal.
- Choose the unit cell shape. Either by writing its cell parameters (first tab) or directly by writing the unit cell vectors (second tab). The button Write Matrix/Write Vectors allows you to pass from one notation to the other.
-
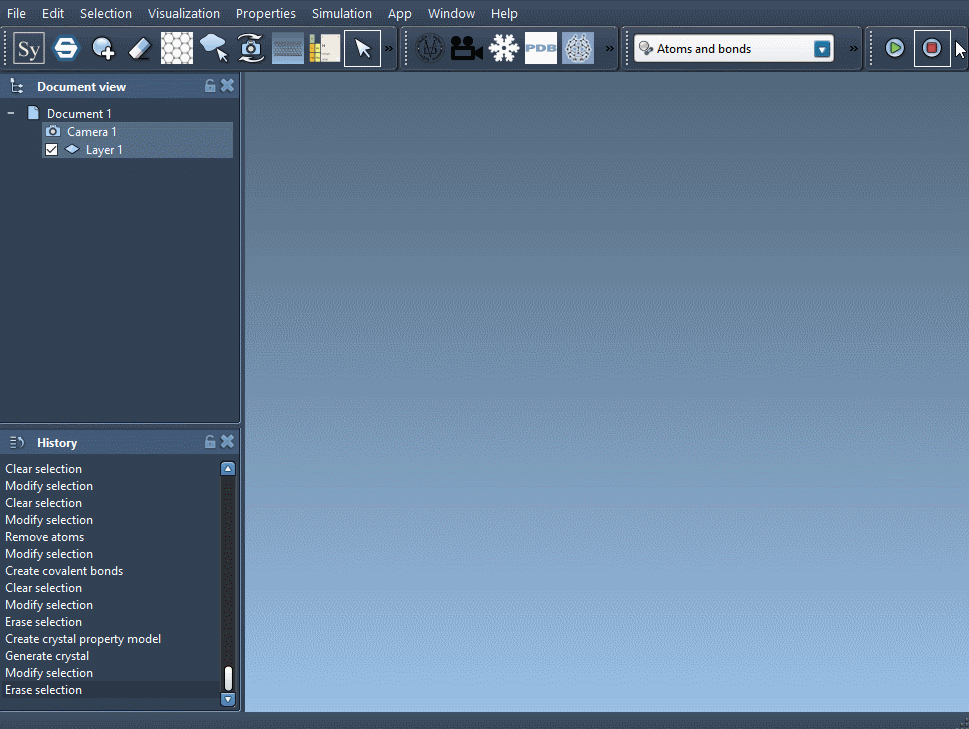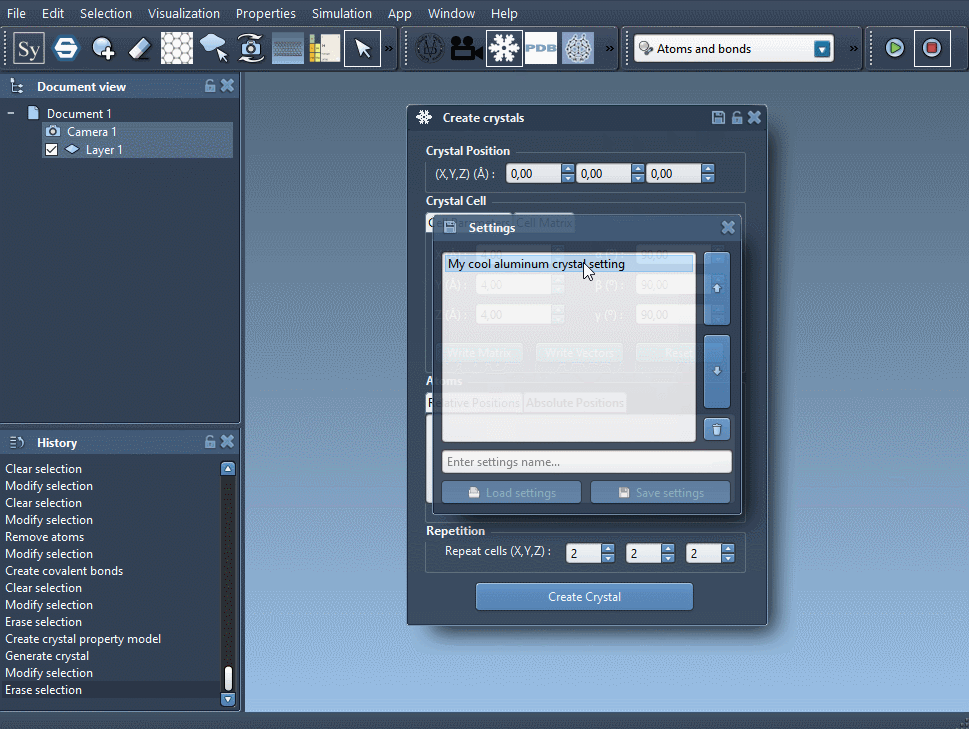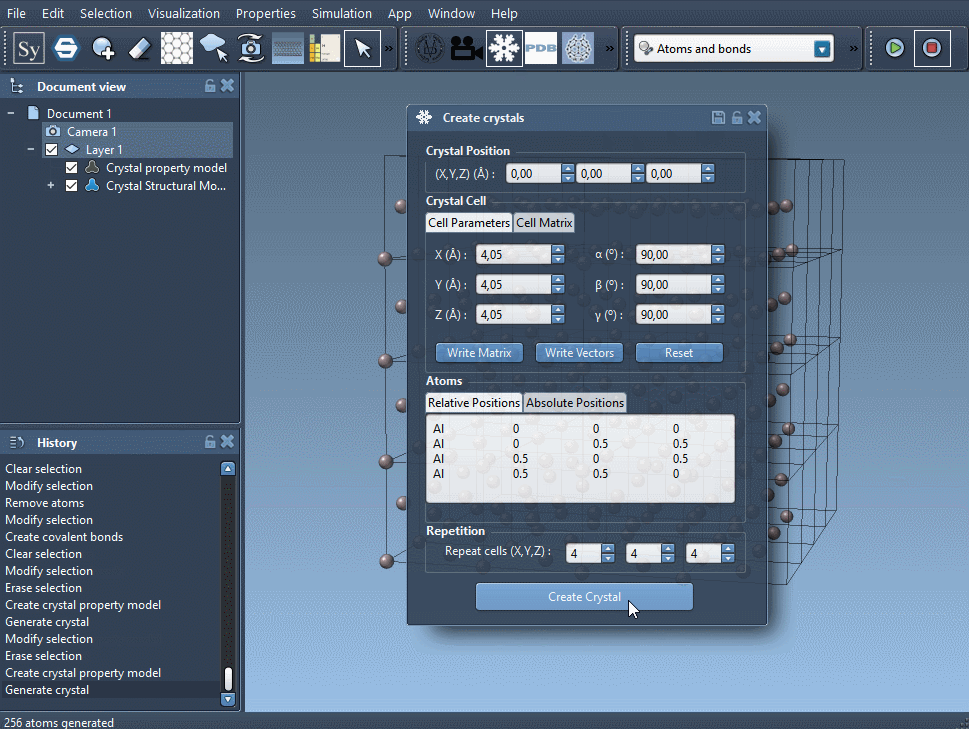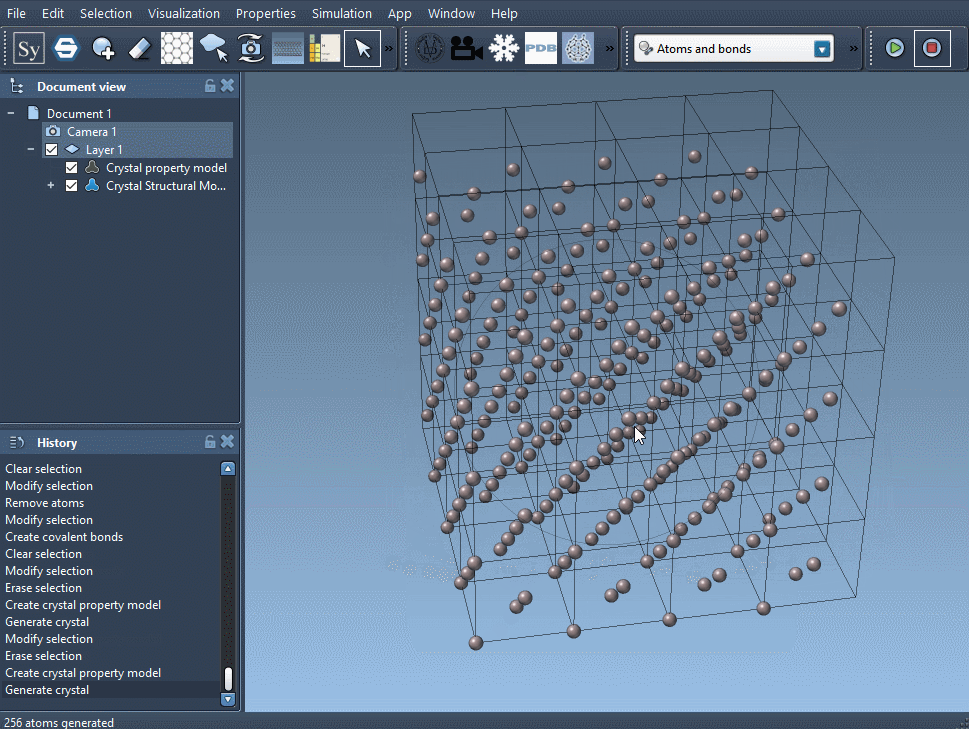
Choose the atoms composing your unit cell. It should be written like this:
<atom1 symbol> <atom1 x-coordinate> <atom1 y-coordinate> <atom1 z-coordinate> <atom2 symbol> <atom2 x-coordinate> <atom2 y-coordinate> <atom2 z-coordinate> <atom3 symbol> <atom3 x-coordinate> <atom3 y-coordinate> <atom3 z-coordinate> ...The atom symbol has to start by its atomic element symbol (C, Al, Kr ...) that can be followed - without space - by other characters to identify it. Depending on the tab you are in, the atom coordinates are in 'relative position', i.e., a proportion of the unit cell vectors X, Y and Z, or in 'absolute position', in angstroms.
-
Choose the number of unit cells you want to generate.
You can also save and load all your crystals by saving their settings ('save' icon on the property window).
Examples#
For a Face-Centered Cubic (FCC) Aluminum crystal, take a cubic unit cell of lattice parameter 4.05 Å, and write in the 'relative position' tab:
Al 0 0 0
Al 0 0.5 0.5
Al 0.5 0 0.5
Al 0.5 0.5 0
If you want a diamond crystal, you can choose a cubic unit cell of lattice parameter 3.60 Å and write in the 'relative position' tab:
C0 0 0 0
C1 0.25 0.25 0.25
C2 0 0.5 0.5
C3 0.5 0 0.5
C4 0.5 0.5 0
C5 0.25 0.75 0.75
C6 0.75 0.25 0.75
C7 0.75 0.75 0.25
Now, try to create your own Sphalerite (SiZ) crystal, and use the SAMSON tool to create all the bonds.
Defects in diamond#
We will see the effect of defects on the structure of diamond:
- Load your diamond crystal (the .cif one) and create the bonds.
- Minimize its structure with the Brenner interaction model.
- Make a copy of the diamond file and open it in a text editor.
- At the end of the new file, instead of
loop_
_atom_site_label
_atom_site_fract_x
_atom_site_fract_y
_atom_site_fract_z
C 0.00000 0.00000 0.00000
copy and paste:
loop_
_atom_site_label
_atom_site_fract_x
_atom_site_fract_y
_atom_site_fract_z
_atom_site_occupancy
C 0.00000 0.00000 0.00000 0.95
The inserted line and the last figure mean our carbon atom has a probability of 0.95 to be present.
- Load your diamond with defects, create the bonds.
- See how the structure changed with defects.
Related tutorials#
- Build carbon nanotube models for another materials-science structure-building workflow.
- UMA Force Field if you want to use machine-learning atomistic models after building a structure.
Next step#
Open the Extensions Tutorials materials-science section when you want more structure-building workflows.
Need Help?#
Have questions or feedback? Feel free to reach out via the Forum, via e-mail, via the Feedback button in SAMSON, or by directly discussing with us.