Covalent and non-covalent protein-ligand docking with the Fitted Suite by Molecular Forecaster#
Use the FITTED Suite SAMSON Extension to run both covalent and non-covalent protein-ligand docking workflows in SAMSON.
The FITTED Suite SAMSON Extension was developed in partnership with Molecular Forecaster and wraps their Fitted Docking software 1, 2.
FITTED stands for Flexibility Induced Through Targeted Evolutionary Description. This fully automated docking software is unique in that it considers the flexibility of macromolecules, the presence of bridging "displaceable" water molecules, covalent functional groups, and proton shifts upon metal coordination. FITTED is based on a genetic algorithm with an emphasis on balancing speed and accuracy. It has excellent scoring functions and accuracy (see third-party studies) and can be applied to metalloenzymes, kinases, nucleic acids, NHRs, GPCRS, etc.
The FITTED Suite SAMSON Extension automates the protein-ligand docking by using the following accessory programs by Molecular Forecaster:
- PREPARE (Protein Rotamers Evaluation and Protonation based on Accurate Residue Energy) automates protein preparation by cleaning up frequent liabilities, optimizing various physicochemical properties, and orienting water molecules.
- PROCESS (Protein Conformational Ensemble System Setup) generates modified protein files for their use with FITTED.
- SMART (Small Molecule Atom-typing and Rotatable Torsion assignment) characterizes small molecules for their use with FITTED and IMPACTS.
- CONVERT (Conformational Optimization of Necessary Virtual Enantiomers, Rotamers, and Tautomers) transforms 2D small molecules into accurate 3D representations.
What you will learn#
In this tutorial, you will learn how to set up and run covalent and non-covalent protein-ligand docking workflows with the FITTED Suite in SAMSON.
Before you start#
- Add the FITTED Suite SAMSON Extension.
- Download the FITTED tutorial archive.
- Keep in mind that the page covers two related workflows, so it is best followed from top to bottom the first time.
Non-covalent docking: 1E2K#
Launch SAMSON and open the 1E2K-A.sam file provided in the archive. To open a file in SAMSON, click on Home > Open or simply drag-and-drop the file in SAMSON. You can also download this file directly in SAMSON from SAMSON Connect - Assets, by clicking on Home > Download and providing the following link: https://www.samson-connect.net/documents/25974716-0526-49b2-81a6-575c6a1a4f46.
This will open a structural model of a thymidine kinase protein (1E2K, chain A) with the (N)-methanocarba-thymidine ligand (TMC 500) bound to it. You should see the following in the Document view:

The Document view is a panel that shows the data graph structure representation of the opened document. In SAMSON, documents are hierarchies of SAMSON nodes (molecules, folders, visual models, cameras, etc.). A document hierarchy is visible in the Document view and its 3D representation in the Viewport (see User guide: Interface for more information). You can read more about documents in User guide: Documents.
Tip
If you have the Document view closed, you can open it via Interface > Document view or via the Ctrl+1 shortcut on Windows and Linux or Cmd+1 on Mac.
Preparation of the system#
There is no preparation needed for the system in the tutorial. The FITTED Suite will automatically deal with water molecules and will use PREPARE to adjust bond order, add hydrogens, generate the possible tautomers, and optimize the H-bond network by an iterative algorithm. But, generally, you would need to check if atoms in the system have alternate locations and remove them if necessary. For that, you can use Home > Prepare. Please refer to the Protein Preparation & Validation tutorial for more information on how to prepare and fix protein systems.
Setup of the system#
Let's now open the FITTED Suite app by selecting it in the Home > Apps > Biology. You can also find it using the Find everything... search box in the top menu of SAMSON - just start typing the app's name.
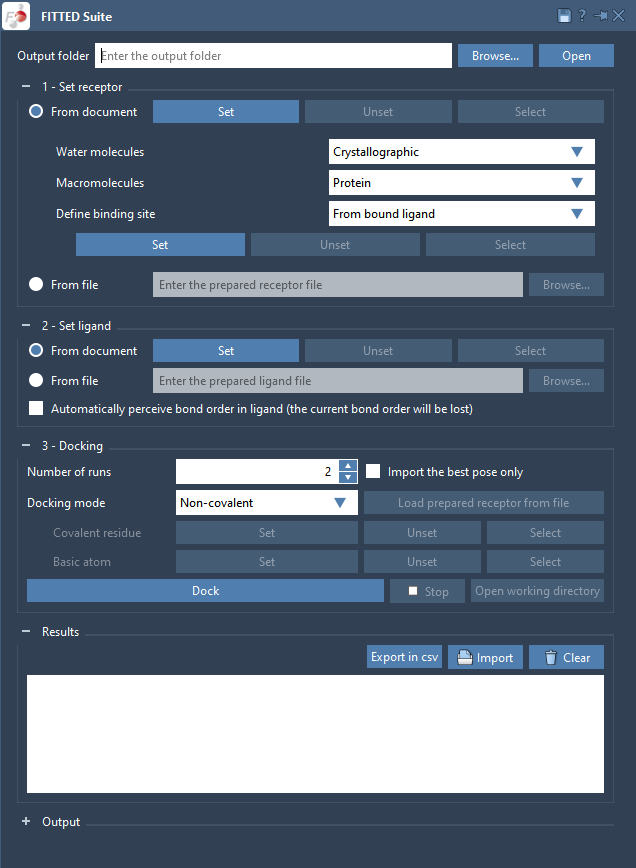
Now, to set up the system we need to define the following:
- a receptor;
- a binding site;
- a ligand.
Setup of the receptor#
Select the 1E2K structural model from the document.

Then in the Set receptor part of the FITTED Suite select From document and click on the corresponding Set button.
Leave the Water molecules parameter to its default value and the Macromolecules parameter to its default value as well (Protein).

Note that if you would like to dock a metalloprotein, DNA, or RNA you should change the Macromolecules parameter accordingly.
Setup of the binding site#
There are three options to specify the binding site:
- From bound ligand - specify the binding site based on an already bound ligand.
- From selection - specify the center of the search grid based on the centroid of the selected atoms.
- From position - specify the center of the search grid using a sphere in the Viewport (the central panel with a 3D representation of the document).
Since in this tutorial we will be performing self-docking (docking of the ligand in its own crystal structure receptor) and we already have a bound ligand (TMC 500), we can specify the binding site based on it.
The TMC 500 bound ligand is part of chain A. You can find it in the document by expanding chain A or simply by typing TMC 500 in the search bar of the Document view. Select the TMC 500 ligand:

For the sake of simplicity, this ligand is also referred to by the TMC 500 group. Double-click on the TMC 500 group in the document to select the ligand.

Now, in the Define binding site part of the FITTED Suite select From bound ligand and click on the corresponding Set button.
If you do not have a bound ligand, you can specify the binding site based on its position by selecting the From position option and moving in the Viewport the sphere representing the center of the binding site.
Setup of the ligand#
Select the TMC 500 ligand. The TMC 500 bound ligand is part of chain A. You can find it in the document by expanding the chain A or simply by typing TMC 500 in the search bar of the Document view. Select the TMC 500 ligand. For the sake of simplicity, the TMC 500 ligand is also referred to by the TMC 500 group - you can simply double-click on the TMC 500 group in the document to select the ligand.
Now, in the Set ligand part of the FITTED Suite select From document and click on the corresponding Set button.

Leave the option to prepare the ligand (perceive bond order and add hydrogens) checked.
Running the docking#
Now we are almost ready to launch the docking. We only need to specify some docking parameters (you can leave them by default):
- Set the number of runs to 2.
- Check the option to import the best pose only if you want to import only the best pose.
- Set the docking mode to Non-covalent.

At the top of the app, you can also select the Output folder where you would like the results to be saved.

Once you have specified the necessary parameters, click on the Dock button. For the system in the tutorial, the docking may take a few minutes - the current stage is shown on this button and the logs can be seen below the results table.
Results#
Once the docking calculations are done, the results should be shown in the results table and automatically loaded in the document. Please note that if you selected to import only the best pose then results only for this pose will be loaded in the table.

You can export the table with results in a CSV file or copy each row separately via its context menu. When selecting a row in the results table it will also select the corresponding pose in the document.
In the document, you should see a processed receptor (1E2K_pro) with added hydrogens and the resulting ligand pose.

Let's now hide the initial structure by unchecking it in the Document view as follows:

Performing further analysis#
SAMSON provides different tools to perform analysis of the results. You can add various visual models, measure distances, compute some parameters, and check for ligand-receptor interactions. Below you will find some examples.
Visualizing#
Let's first add some visual models for the system. You can learn more about the visualization in SAMSON thanks to the interactive tutorials (Help > Tutorials) or to the User guide: Visualizing.
First, in the Document view, select the processed receptor (1E2K_pro) structural model and use the Context Toolbar or the Visualization menu to add a Ribbons structure. This should add a new secondary structure visual model to the document. Now you can hide the 1E2K_pro structural model by unchecking it in the Document view.
Tip
If you want to hide a visual model, simply uncheck it in the Document view; if you want to remove it then right-click on it and choose Erase from the context menu.

Let's now add labels and a licorice visual for residues surrounding the ligand. First, we need to select residues surrounding the ligand. For that, select the resulting ligand - 1E2K_log.mol2_DockingRun_* - and click on Select > Biology > Binding sites and set the parameters as in the image below and click OK. This will select the residues surrounding the currently selected ligand.

You can save this selection in a group by clicking on the Current selection button in the Document view and clicking on Create group in the context menu or on Select > Group. Name the group e.g. as "Binding site residues".

Let's now add a licorice visual for these residues. While having these residues selected, go to the Visualization menu and click on the Licorice button - this will add a licorice visual model for the selected residues.
If you want, you can colorize carbons in the receptor based on e.g. the residue sequence number. For that, select the receptor (1E2K_pro) and go to Select > Atoms > Carbons.

This will select all carbons in the current selection. Let's now colorize them by the residue sequence number such that their color would correspond to the default style of secondary structure colorization. For that, choose from the Visualization menu > Material > Per attribute menu the Residue index option. This should colorize both the carbons in the receptor's structural model and in the added licorice visual model since the licorice visual model doesn't have a specific color scheme applied to it.
Now let's hide the receptor's structural model by unchecking it in the document if you didn't already do it before. In the end, you should see something like in the image below:

Please note that you can modify the appearance of visual models using the Inspector.
If you want to learn more about visual models in SAMSON, please follow interactive tutorials in SAMSON (Help > Tutorials) or the User guide: Visualizing tutorial.
Protein-Ligand Interaction Analyzer#
The Protein-Ligand Interaction Analyzer SAMSON Extension allows for computing the contact area, H-bonds between a ligand and a receptor, ligand surrounding residues, and some other parameters. Check out its other tabs for more functionality.
Open the Protein-ligand Interaction Analyzer from the Home > Apps > Biology or simply by searching by its name in the Find everything... search bar in the top menu of SAMSON.
Select the resulting receptor (1E2K_pro) and click on Set for Receptor.
Choose the Single ligand option from the list. Select the resulting ligand (1E2K_log.mol2_DockingRun_*) and click on Set for Ligand.
Now, click Analyze.

Hydrogen bonds#
The Hydrogen Bond Finder extension allows one to find and visualize hydrogen bonds (H-bonds) inside a molecule or between molecules, e.g. between a ligand and a receptor. To find H-bonds between a ligand and a receptor, select the second option ("... in the current selection and the system"), then select the resulting receptor (1E2K_pro) and click on the Set button, modify parameters if necessary (you can later modify them in the created H-bond visual model using the Inspector), then select the resulting ligand (1E2K_log.mol2_DockingRun_*) and click on the Add hydrogen bond visual model button.
This will add a new H-bond visual model to the document. If you want to modify the H-bond detection parameters, right-click on the newly created H-bond visual model and choose Inspect from the context menu - this will open the Inspector in which you can modify parameters.

Covalent docking: 5MAJ#
Please make sure to go through the Non-covalent docking: 1E2K part of the tutorial first, since in this section we will be omitting some of the descriptions already mentioned in the section above.
Launch SAMSON and open the 5MAJ.sam file provided in the archive. To open a file in SAMSON, click on Home > Open or simply drag-and-drop the file in SAMSON. You can also download this file directly in SAMSON from SAMSON Connect - Assets, by clicking on Home > Download and providing the following link: https://www.samson-connect.net/documents/c5a6fc63-0391-4f88-bd60-fcc1313e47fa.
This will open a structural model of the human cathepsin L protein (5MAJ) with a 7KH ligand (7KH 301) covalently bound to it. You should see the following in the Document view:

Tip
If you have the Document view closed, you can open it via Interface > Document view or via the Ctrl+1 shortcut on Windows and Linux or Cmd+1 on Mac.
Preparation of the system#
There is no preparation needed for the system in the tutorial. The FITTED Suite will automatically deal with water molecules and will use PREPARE to adjust bond order, add hydrogens, generate the possible tautomers, and optimize the H-bond network by an iterative algorithm. But, generally, you would need to check if atoms in the system have alternate locations and remove them if necessary, and fix missing residues and heavy atoms. Please refer to the Protein Preparation & Validation tutorial for more information on how to prepare and fix protein systems.
Modifying bond order and hybridization#
For some protein-ligand complexes where a ligand is already covalently bound to the protein, it might be necessary to modify some bond orders for an atom covalently bound to a receptor and specify the hybridization for some atoms in the ligand. This step is not necessary if you are docking a non-bound ligand.
In the tutorial example, this has already been done, but we will describe here what has been done and how to do it. For convenience, the tutorial example has some saved groups in the document that link to a ligand and some other nodes. Let's zoom in on the part of the 7KH 301 ligand that needs a change in hybridization. Select the C8 atom in the 7KH 301 ligand (expand chain A in the document and scroll down or type 7KH 301 in the search bar) or simply double-click on the C8 in 7KH 301 group in the document - this will select the C8 atom in it - and click Shift+Space to zoom on the selection:

Here we have changed the bond order for the C8-N7 bond to the triple bond as the ligand is nitrile when unbound. This can be done either using the Edit > Edit bonds editor or by selecting the bond and changing the corresponding parameters in the Inspector.

Then we have set the hybridization for C8 and N7 atoms in the 7KH 301 ligand to SP by selecting them and modifying the corresponding parameters in the Inspector.

Setup of the system#
Let's now open the FITTED Suite app by selecting it in the Home > Apps > Biology. You can also find it using the Find everything... search box in the top menu of SAMSON - just start typing the app's name.
Now to set up the system we need to define the following:
- a receptor;
- a binding site;
- a ligand.
Setup of the receptor#
Select the 5MAJ structural model from the document and then in the Set receptor part of the FITTED Suite select From document and click on the corresponding Set button.
Leave the Water molecules parameter to its default value and the Macromolecules parameter to its default value as well (Protein). Note that if you would like to dock a metalloprotein, DNA, or RNA you should change the Macromolecules parameter accordingly.
Setup of the binding site#
Since in this tutorial we will be doing a self-docking and we already have a bound ligand (7KH 301), we can specify the binding site based on it.
The 7KH 301 bound ligand is part of chain A. You can find it in the document by expanding chain A or simply by typing 7KH 301 in the search bar of the Document view. Select the 7KH 301 ligand:

For the sake of simplicity, this ligand is also referred to by the 7KH 301 ligand group. Double-click on the 7KH 301 ligand group in the document to select the ligand.

Now, in the Define binding site part of the FITTED Suite select From bound ligand and click on the corresponding Set button.
If you do not have a bound ligand, you can specify the binding site based on its position by selecting the From position option and moving in the Viewport the sphere representing the center of the binding site.
Setup of the ligand#
Select the 7KH 301 ligand. The 7KH 301 bound ligand is part of chain A. You can find it in the document by expanding chain A or simply by typing 7KH 301 in the search bar of the Document view. For the sake of simplicity, the 7KH 301 ligand is also referred to by the 7KH 301 ligand group. Double-click on the 7KH 301 ligand group in the document to select the ligand. Now, in the Set ligand part of the FITTED Suite, select From document and click on the corresponding Set button.
Uncheck the option to prepare the ligand (perceive bond order and add hydrogens) since it has a modified bond order and hybridization.

Running the docking#
Now we are almost ready to launch the docking. We only need to specify some parameters for covalent docking:
- Set the number of runs to 2.
- Check to import the best pose only if you want to import only the best pose.
- Set the docking mode to Covalent only.

Now we need to specify a covalent residue and a basic atom, the last one is optional.
A covalent residue is a residue with which a covalent inhibitor will react. You can specify either the residue or an atom in it and the whole residue will be set automatically for you.
In this protein-ligand complex, it is the sulfur atom (SG) in the CYS 25 residue with which the 7KH 301 ligand covalently binds. The CYS 25 residue is part of chain A. You can find it in the document by expanding chain A or simply by typing CYS 25 in the search bar of the Document view. Select the CYS 25 residue.
Tip
Click Shift+Space to zoom on the selection.

For convenience, the CYS 25 residue is also referred to by the CYS 25 group. You can simply double-click on the CYS 25 group - this should select the CYS 25 residue - and then click on the Set button for Covalent residue.

The setting of a basic atom is optional - it should be an atom in a residue adjacent to the covalent residue. Example: nitrogen in an adjacent histidine residue. We will specify the ND1 nitrogen atom from the HIS 163 residue which is adjacent to the covalent residue. The HIS 163 residue is part of chain A. You can find it in the document by expanding chain A and then expanding the residue and its side chain to select the ND1 nitrogen atom. You can also find it by typing "ND1" in "HIS 163" (note the quotes when searching by name) in the search bar of the Document view. Select the ND1 atom.

For convenience, the tutorial example has a group called ND1 in HIS 163 which refers to this atom. In the document, double-click on the ND1 in HIS 163 group - this should select the ND1 atom in the HIS 163 residue.

Once you have the basic atom selected, click on the Set button for Basic atom.
At the top of the app, you can also select the Output folder where you would like the results to be saved.
Once you have specified the necessary parameters, click on the Dock button. For the system in the tutorial, the docking may take a few minutes - the current stage is shown on this button and the logs can be seen below the results table.
Results#
Once the docking calculations are done, the results should be shown in the results table and automatically loaded in the document.
You can export the table with results in a CSV file or copy each row separately via its context menu. When selecting a row in the results table it will also select the corresponding pose in the document.
In the document, you should see a processed receptor (5MAJ_pro) with added hydrogens and the resulting ligand pose. In the case of the covalent docking, the resulting pose will also contain a part of the residue to which it is covalently docked. Let's now hide the initial structure by unchecking it in the Document view and the labels associated with the initial structure by unchecking the Labels folder as follows:

For visualization purposes, let's now add a secondary structure visual model for the receptor. In the Document view, select the processed receptor (5MAJ_pro) structural model and then click on Visualization > Visual model > Ribbons. Now you can hide the 5MAJ_pro structural model by unchecking it in the Document view. You can zoom in on the ligand by selecting it and pressing Shift+Space. You can see that the ligand is covalently bound to the sulfur atom from the CYS 25 residue.

Please check out the Performing further analysis section for more information on protein-ligand interaction analysis.
If you want to learn more about visual models in SAMSON, please follow interactive tutorials in SAMSON (Help > Tutorials) or the User guide: Visualizing tutorial.
That's all, thank you for completing this tutorial on the FITTED Suite SAMSON Extension.
Related tutorials#
- Protein preparation and validation for preparing protein inputs before docking.
- Dock ligands and libraries with AutoDock Vina Extended for another protein-ligand docking route.
- Strain Explorer for analyzing ligand strain in generated or docked conformations.
Next step#
Compare the predicted binding modes, then continue with interaction analysis, analogue generation, or simulation of promising complexes.
Video tutorial#
Learn more about the Fitted Suite from this video tutorial: Protein-ligand docking with the FITTED Suite and the SAMSON molecular design platform.
Need Help?#
Have questions or feedback? Feel free to reach out via the Forum, via e-mail, via the Feedback button in SAMSON, or by directly discussing with us.
References#
-
Moitessier N., Pottel J., Therrien E., Englebienne P., Liu Z., Tomberg A., Corbeil C.R. Medicinal Chemistry Projects Requiring Imaginative Structure-Based Drug Design Methods. Accounts of Chemical Research (2016), 49 (9), 1646-1657 ↩
-
Therrien E., Englebienne P., Arrowsmith A.G., Mendoza-Sanchez R., Corbeil C.R., Weill N., Campagna-Slater V., Moitessier N. Integrating medicinal chemistry, organic/combinatorial chemistry, and computational chemistry for the discovery of selective estrogen receptor modulators with FORECASTER, a novel platform for drug discovery. Journal of Chemical Information and Modeling (2012), 52, 210-224 ↩