Create molecular boxes with Molecular Box Builder#
The Molecular Box Builder extension for SAMSON allows you to fill a 3D box with molecules - you can build solvent boxes, lipid layers, and custom molecular assemblies.
Use this tutorial when you want to populate a box with repeated molecules, estimate how many copies fit, or quickly build a simple solvent or membrane-like environment.
The Molecular Box Builder app can be found in the Home > Apps > Assembly or via the Find everything... (Shift+E) in the top menu of SAMSON.
What you will learn#
In this tutorial, you will learn how to fill a 3D box with molecules in SAMSON using Molecular Box Builder.
Before you start#
- Log into SAMSON Connect.
- Visit the Molecular Box Builder Extension page and click Add.
- Restart SAMSON - your extension will be ready to use.
- Keep the molecule you want to replicate open in the document so you can set it directly from the selection.
Launch the App#
Access Molecular Box Builder via Home > Apps > Assembly > Molecular Box Builder.
Or use Find everything... (Shift+E) at the top of SAMSON and search "Molecular Box Builder".
Step 1 - Set the Molecule to Insert#
- Select a molecule or molecular system with which you would like to populate a box in the Document view or Viewport. This can be one or several molecules, structural models, structural groups, or residues, or even just atoms.
- Click Set in the app.
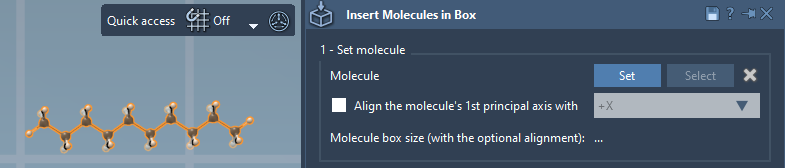
Tip
Click Select in the app to highlight the structure.
Click X in the app to clear the currently set molecule.
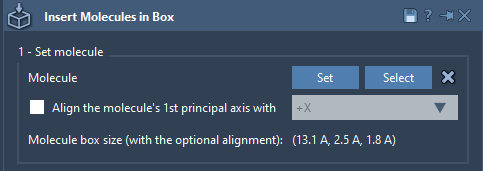
Optional: Define Orientation#
You can specify how molecules should be oriented when added:
- No alignment - use original orientation.
- Align to +X / -X / +Y / -Y / +Z / -Z - align the molecule's first principal axis (the first eigenvector of the moment of inertia) with the selected direction.
Note
You can always move and rotate the system in SAMSON using the Move editors.
SAMSON automatically calculates the bounding box of the molecule based on the chosen alignment.
Step 2 - Define the Box#
- Enter box dimensions
(X, Y, Z). - Optional: to center the box around the origin or a specific point, check Center and adjust the center coordinates. By default, the box is positioned in such a way that its corner is aligned with the origin.
- Optional: specify the margin between inserted molecules (positive or negative). By default, molecules will be added in the box such that there will be no clashes between them.
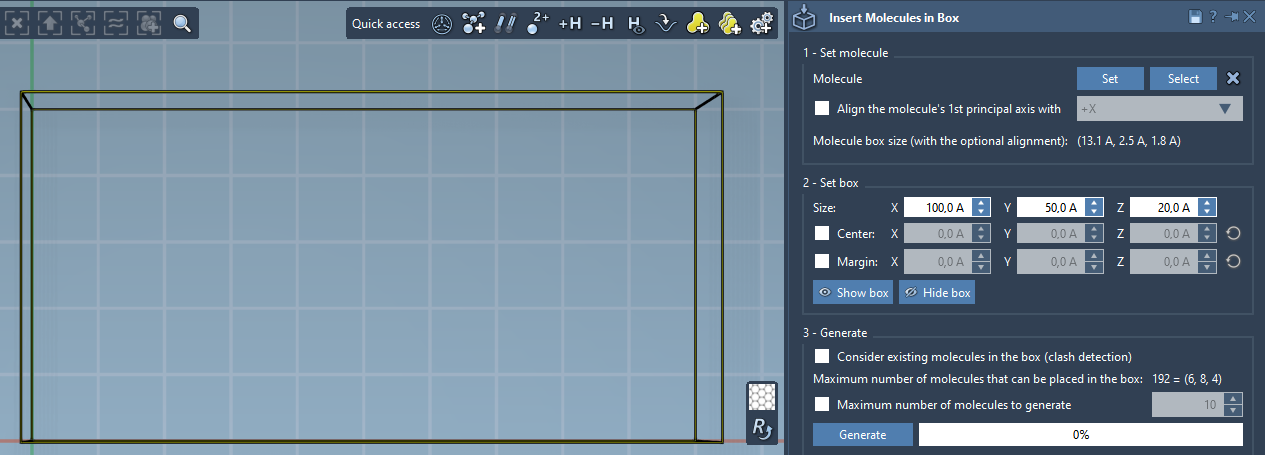
Molecular Box Builder predicts how many molecules can fit along each axis and their total number (shown in the Generate part). Adjusting the box size or molecule's margin updates this prediction.
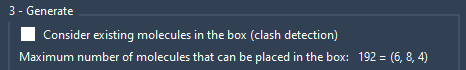
You can toggle visibility of the box as needed by pressing the corresponding buttons in the app.
Step 3 - Generate the System#
To cap the total number of inserted molecules, check Maximum number of molecules to generate and specify a limit. Otherwise, it will place as many molecules as possible in the box, which should be equivalent to the predicted number.
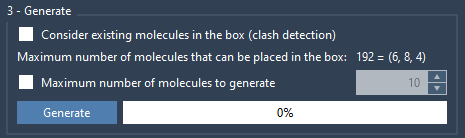
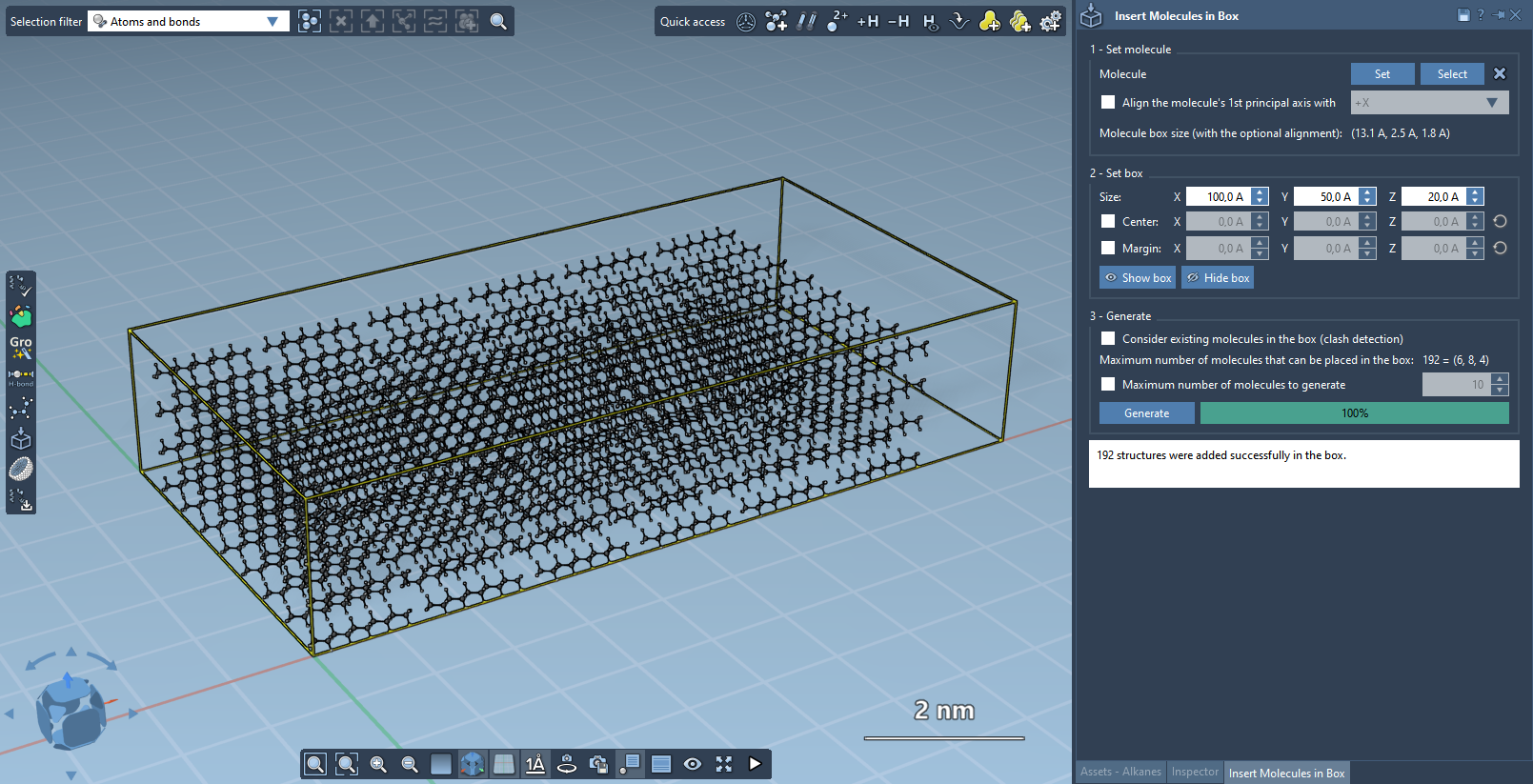
Click Generate. Depending on the size of the system, the generation should take a few seconds.
Result: a filled box populated with non-overlapping copies of the selected molecule.
Build a Lipid Layer Around a Protein#
You can also use Molecular Box Builder to generate lipid membranes around proteins.
Tip
You can also use the PDBFixer extension to build a membrane (from a list of available lipids) around a protein with water and neutralize the system with specified ions.
Let's, for example, build a single layer of lipids around the copper-transporting PIB-ATPase 4BBJ.
Step 1 - Align the Protein#
First, we need to reorient the protein:
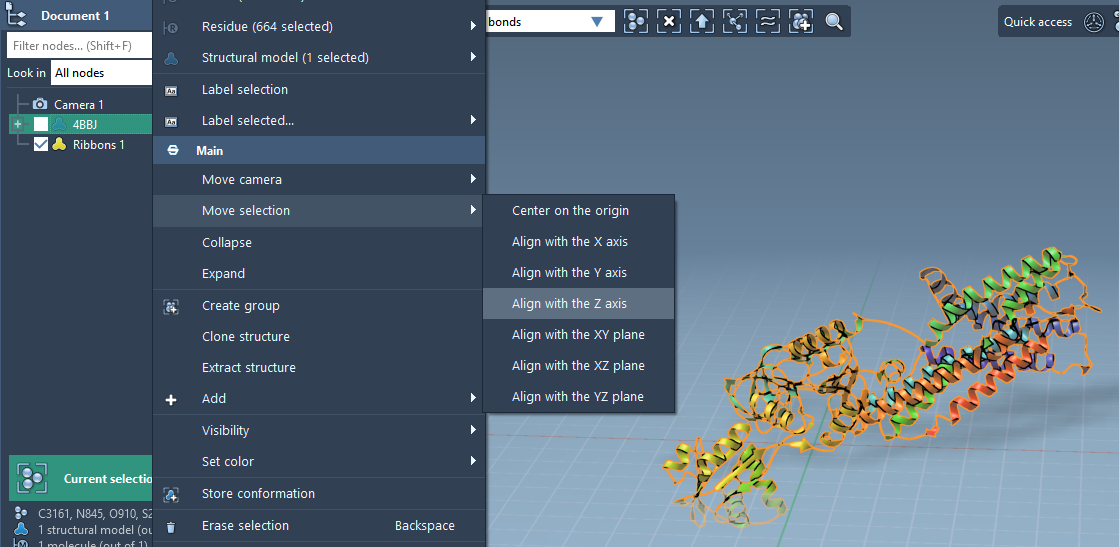
- Right-click the protein in Document view.
- In the context menu, choose: Move selection > Align with Z axis.
- Then: Move selection > Center on the origin.
Step 2 - Set the Lipid Molecule#
- Import a lipid molecule.
- Select it and click Set.
- Align its principal axis to the
+Zaxis.
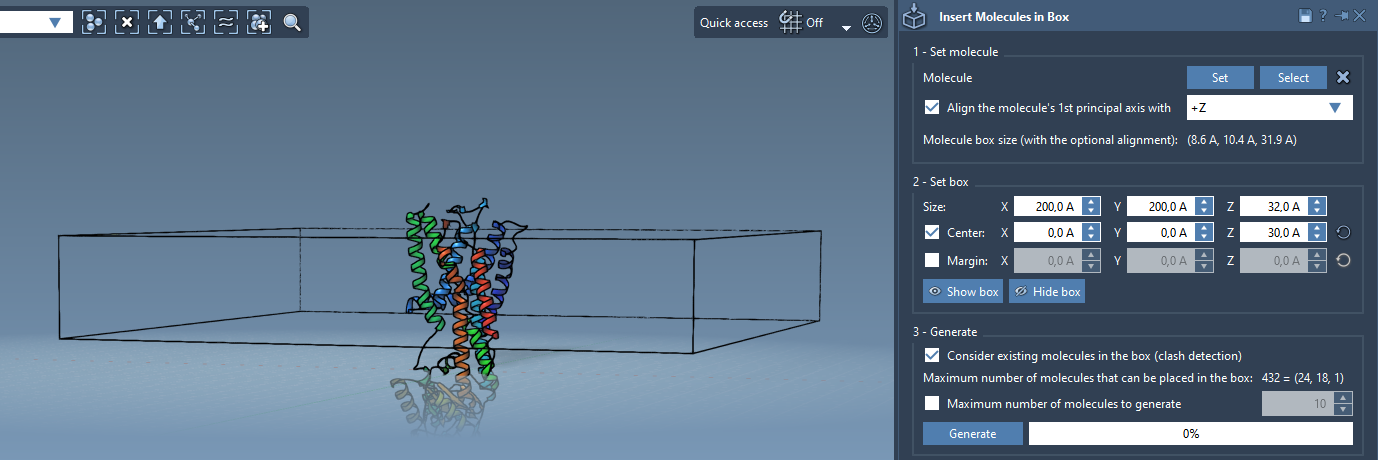
Step 3 - Define the Box#
- Center the box around the protein.
- Adjust the size to contain a single lipid layer.
- Specify the margin between the inserted molecules if needed.
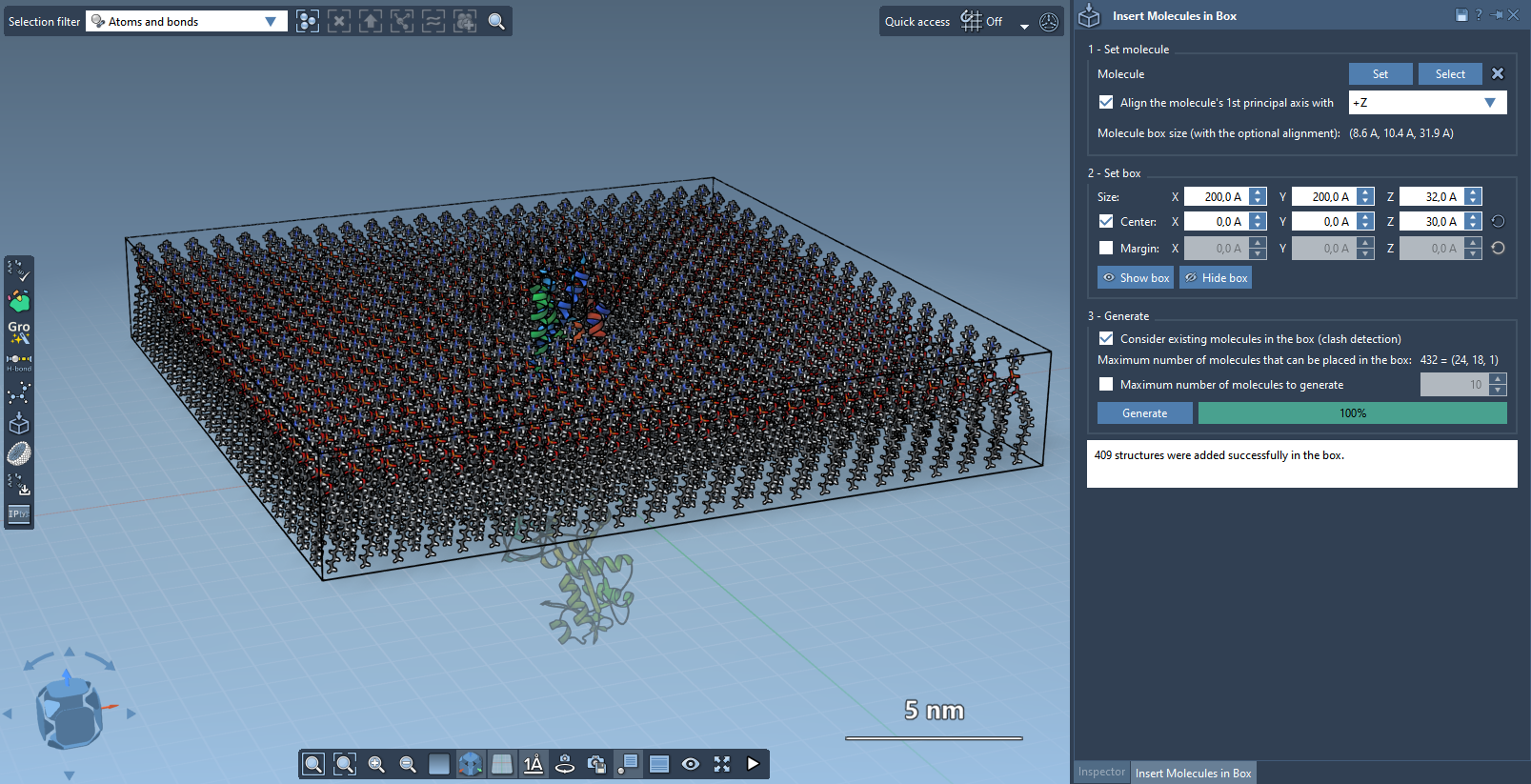
Step 4 - Generate#
- Enable Consider existing molecules in the box so lipids are placed only in available space.
- Click Generate.
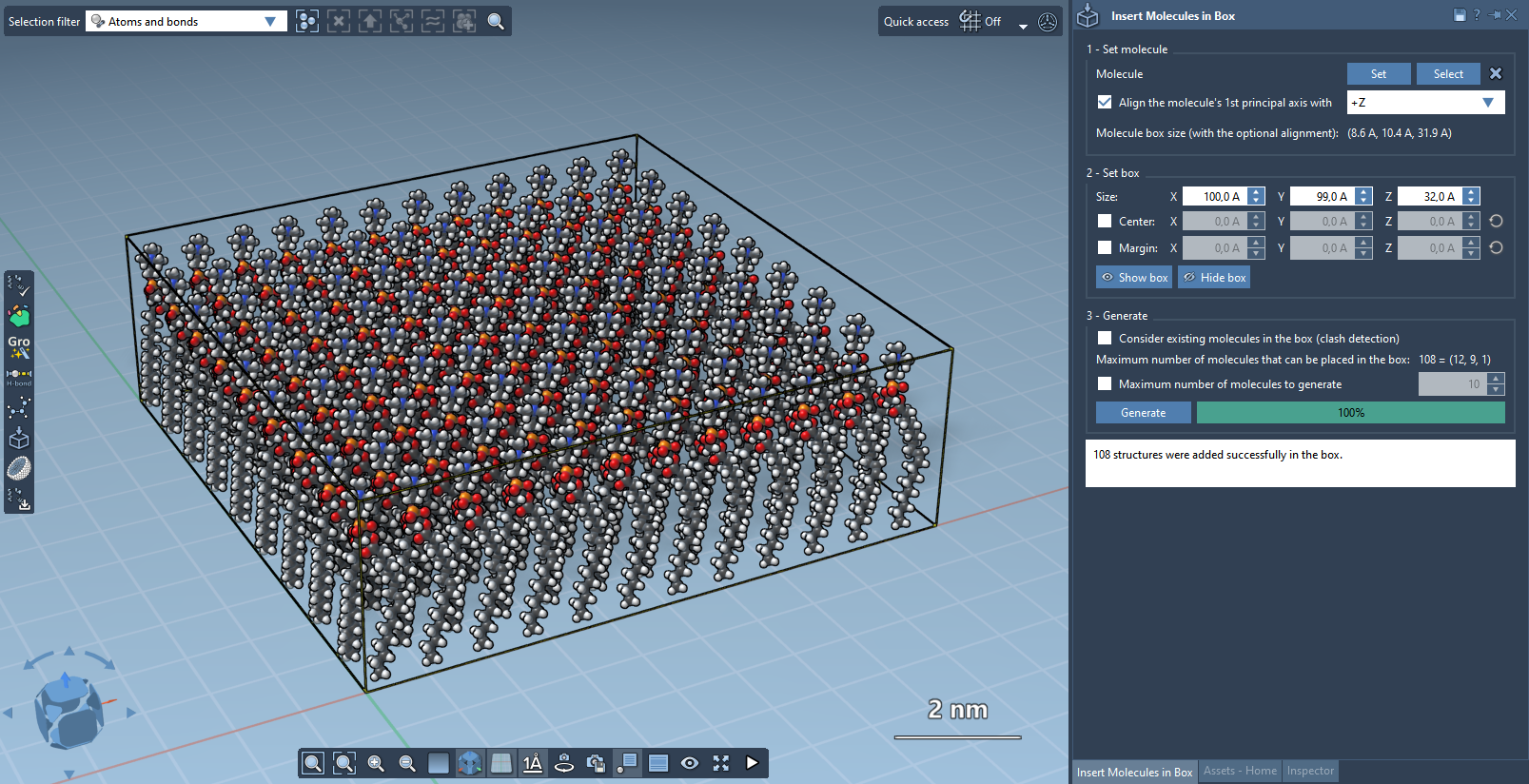
Now you should have a single layer of lipids around the protein:
Optional: Create a Lipid Bilayer#
- Add the first layer with
+Zalignment. - Shift the box center in the
Z-direction. - Add the second layer with
-Zalignment.
Now you have a full lipid bilayer.
Next Steps#
Minimize, equilibrate, and simulate the system using, for example, GROMACS Wizard (see the GROMACS Wizard tutorial).
Related tutorials#
- GROMACS Wizard tutorials for preparing, minimizing, equilibrating, and simulating the generated system.
- Step 1: Prepare with GROMACS Wizard for turning the generated structure into simulation files.
- UMA Force Field for machine-learning based atomistic evaluation.
Need Help?#
Have questions or feedback? Feel free to reach out via the Forum, via e-mail, via the Feedback button in SAMSON, or by directly discussing with us.