Manage and transform SMILES data with the SMILES Manager#
Use the SMILES Manager in SAMSON to import SMILES datasets, review 2D depictions, edit entries, and create 3D-ready structures. The extension is based on RDKit, a widely used open-source cheminformatics toolkit.
This tutorial covers the Manage SMILES and Replace fragments tabs. For the third tab, see the separate Positional Analogue Scanning tutorial. In each section, the SMILES codes are displayed in a table together with names and generated 2D depictions, so you can move quickly from string manipulation to structure generation.
What you will learn#
In this tutorial, you will learn how to import, manage, and transform SMILES data in SAMSON with the RDKit-based SMILES Manager.
Before you start#
- Add the SMILES Manager extension from SAMSON Connect - Marketplace.
- Prepare one or more SMILES strings, or a
.smi/.txtfile, if you want to follow along with your own data. - Use this page when you want to organize or edit a SMILES collection; use the companion tutorial when you want to generate analogue series.
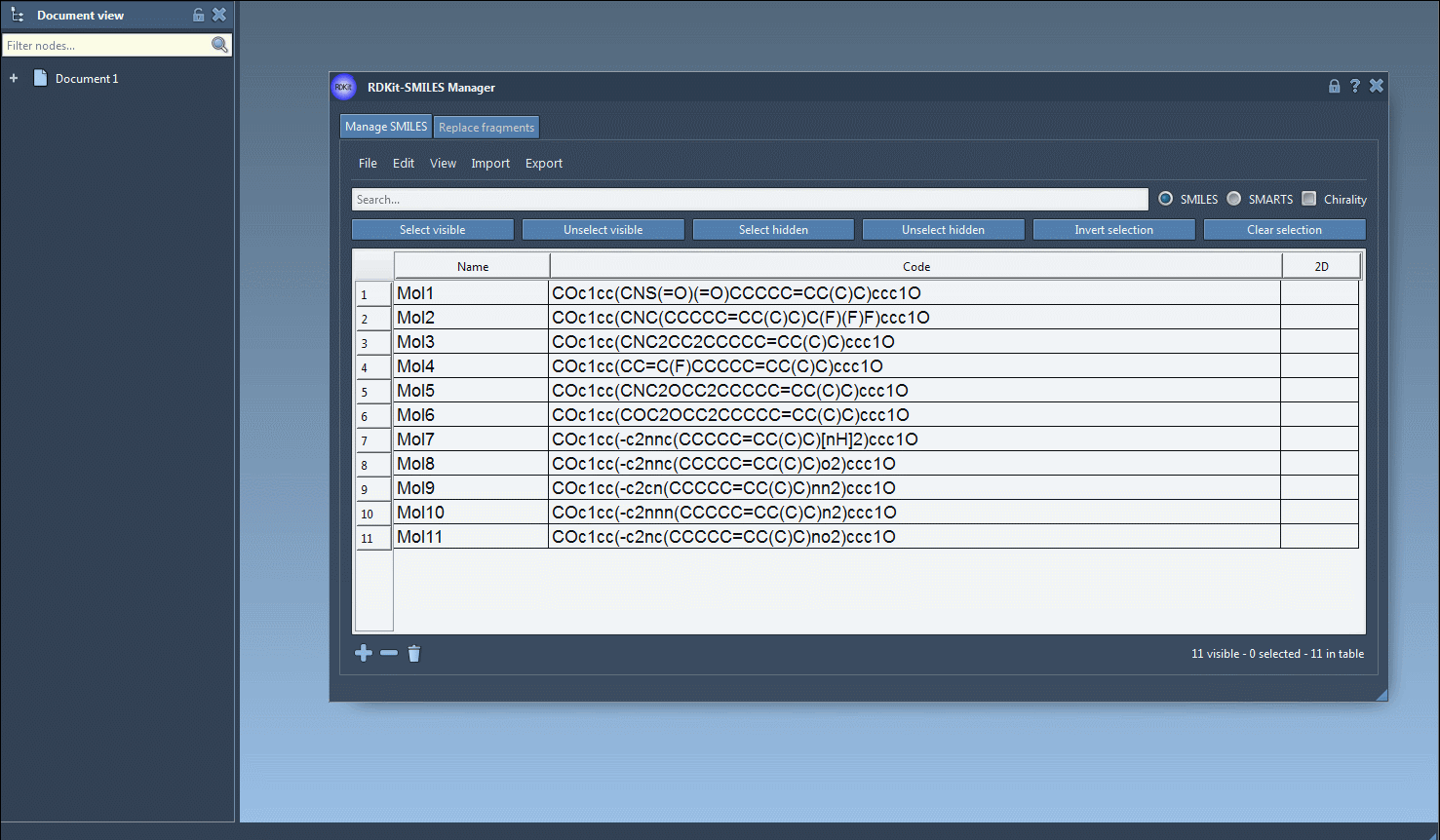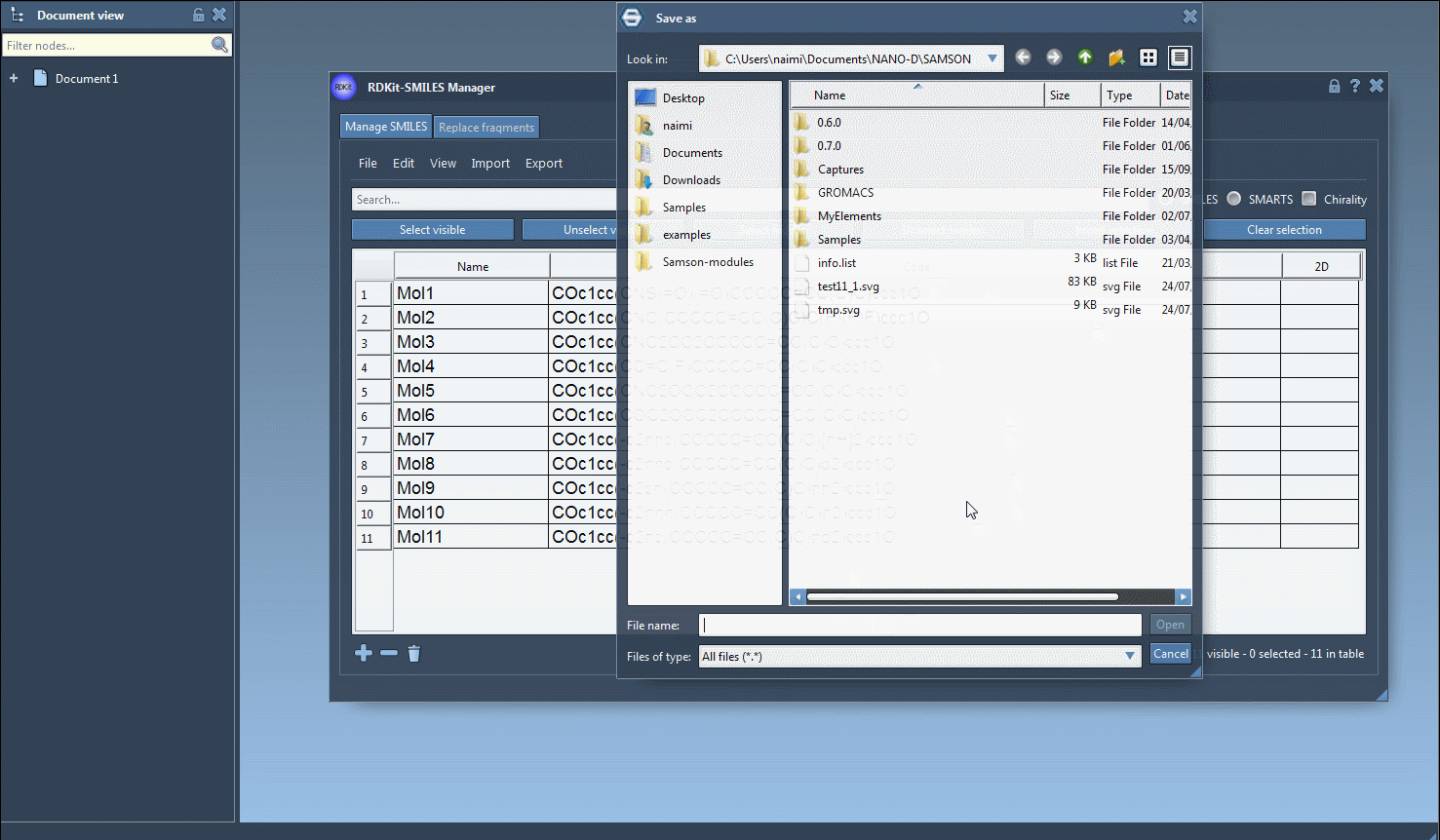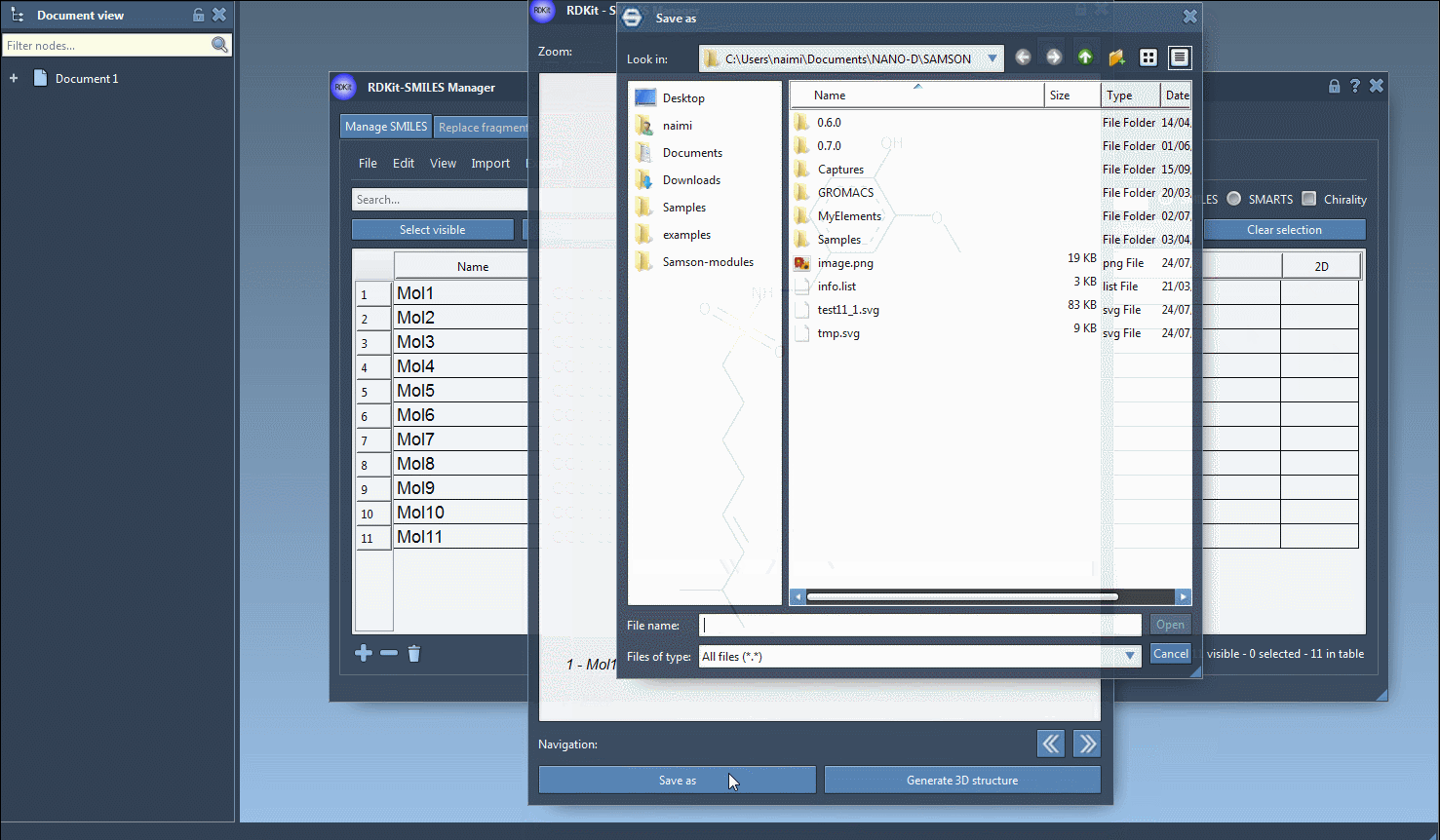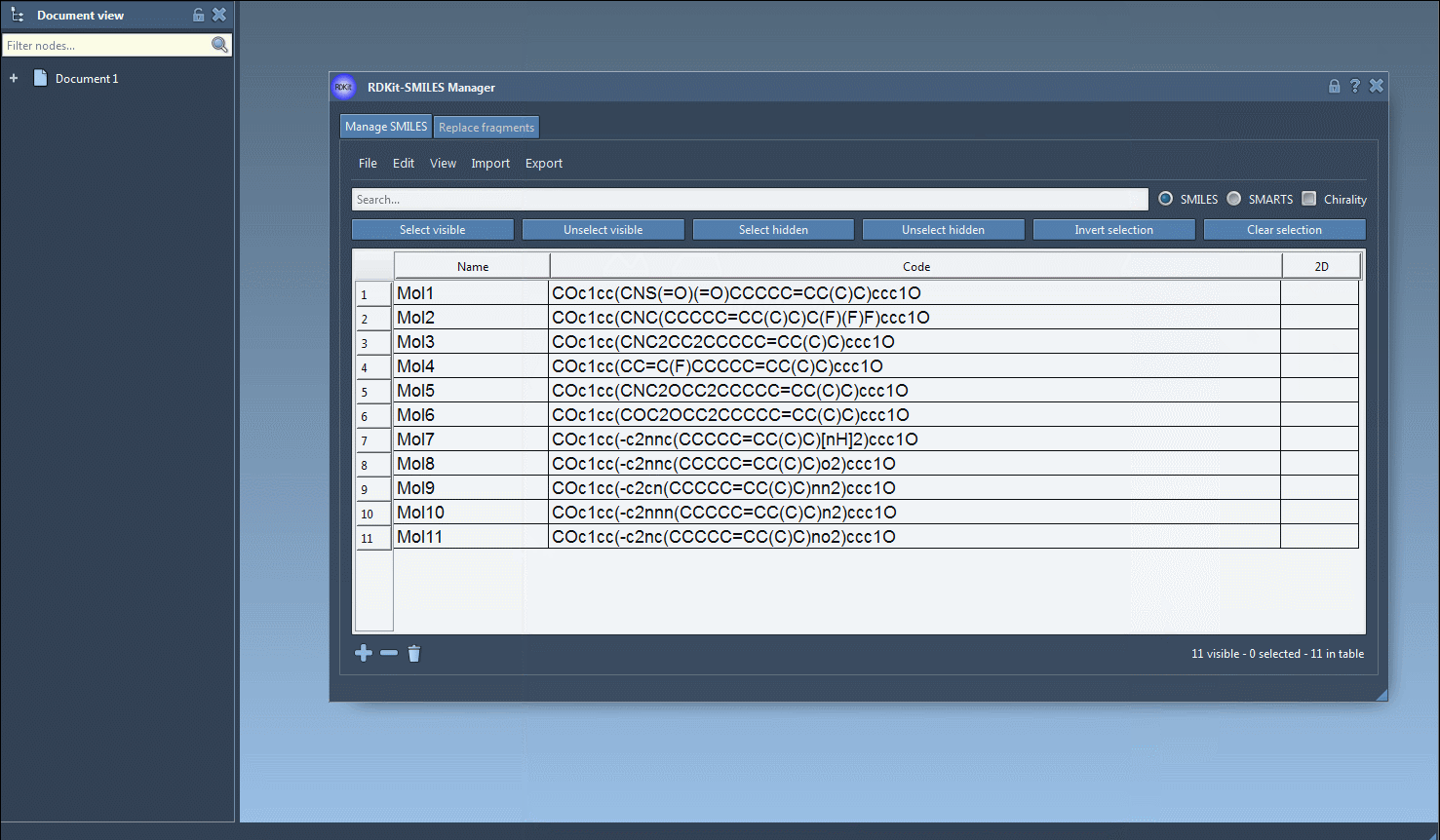
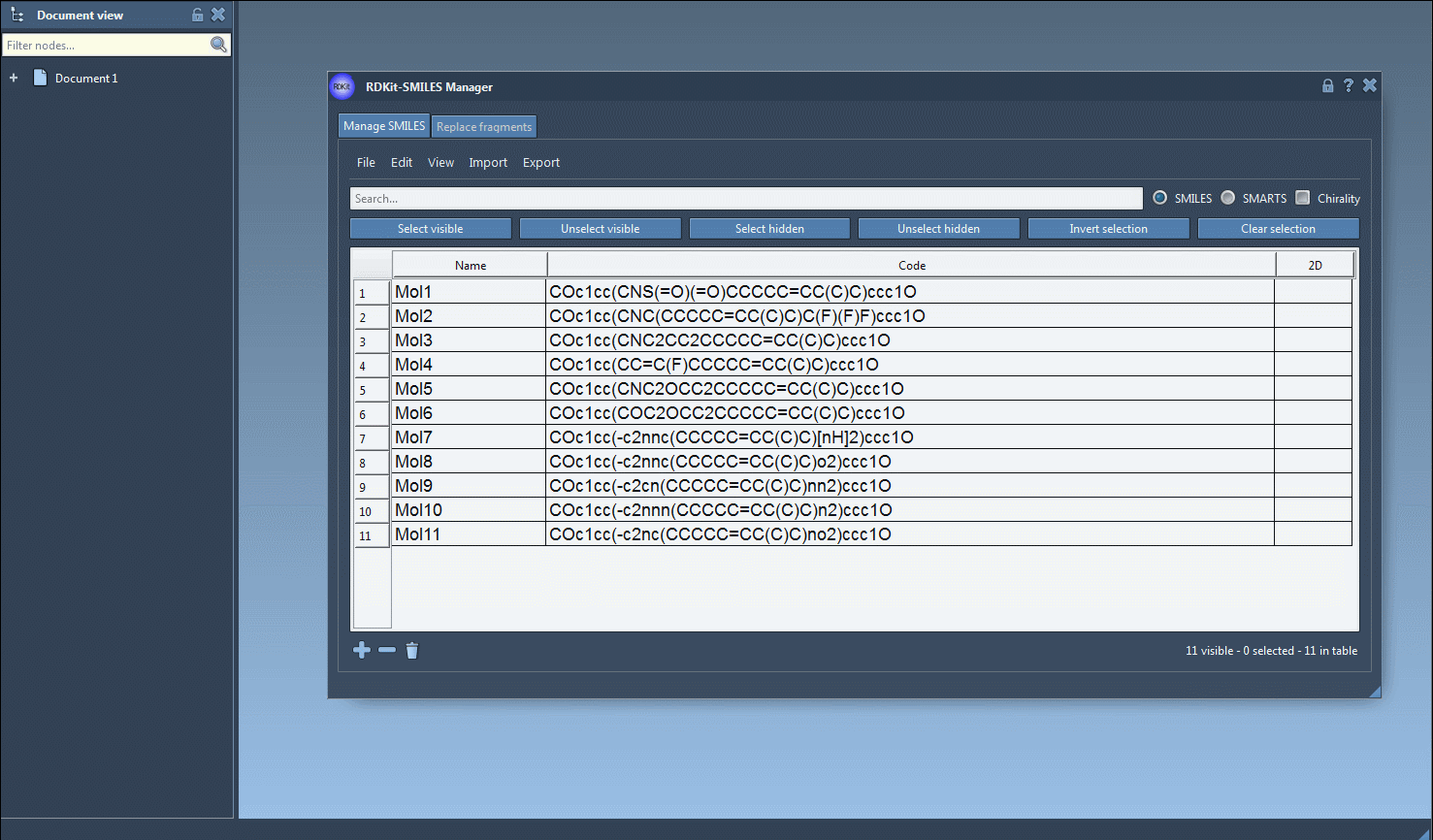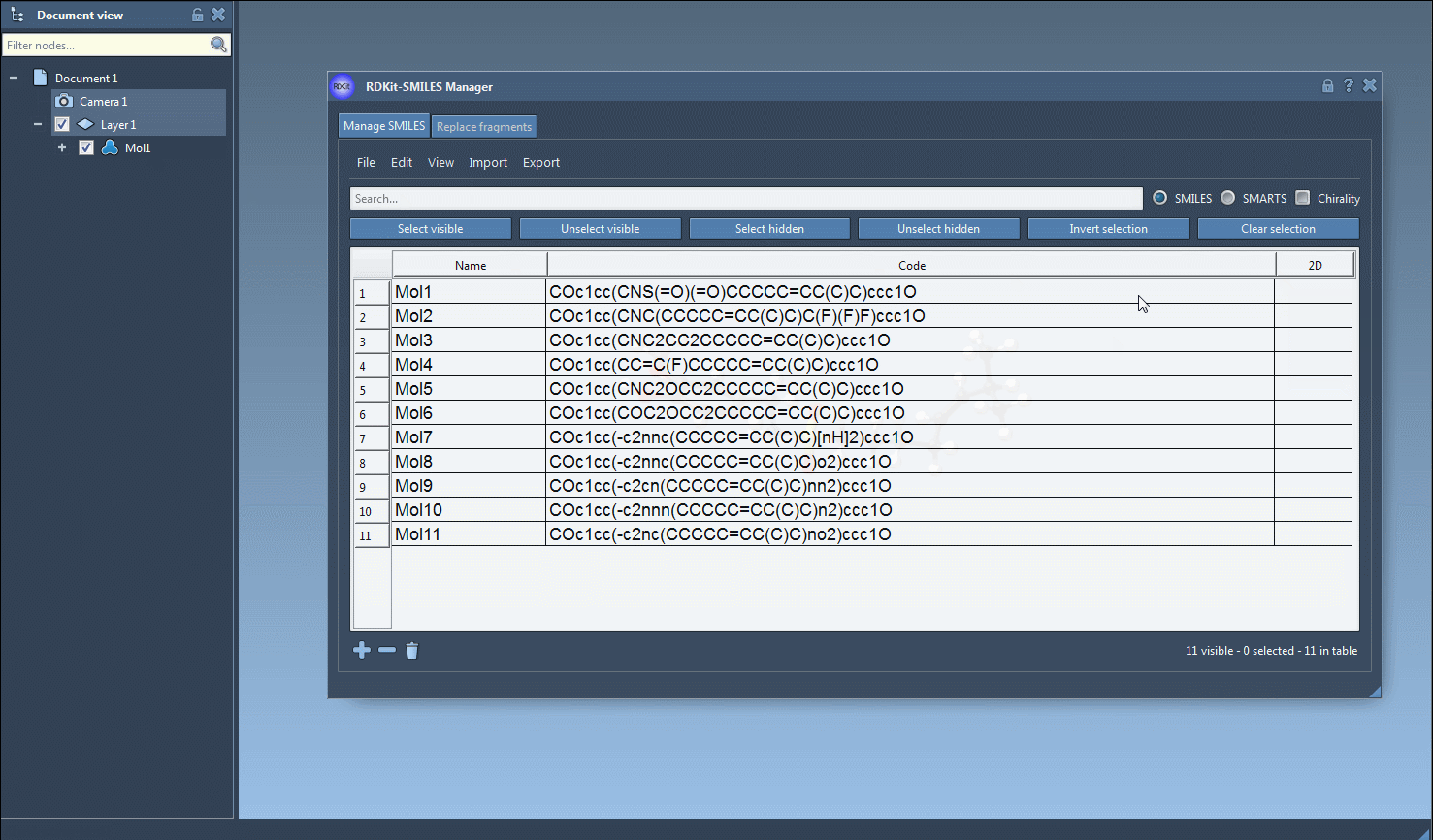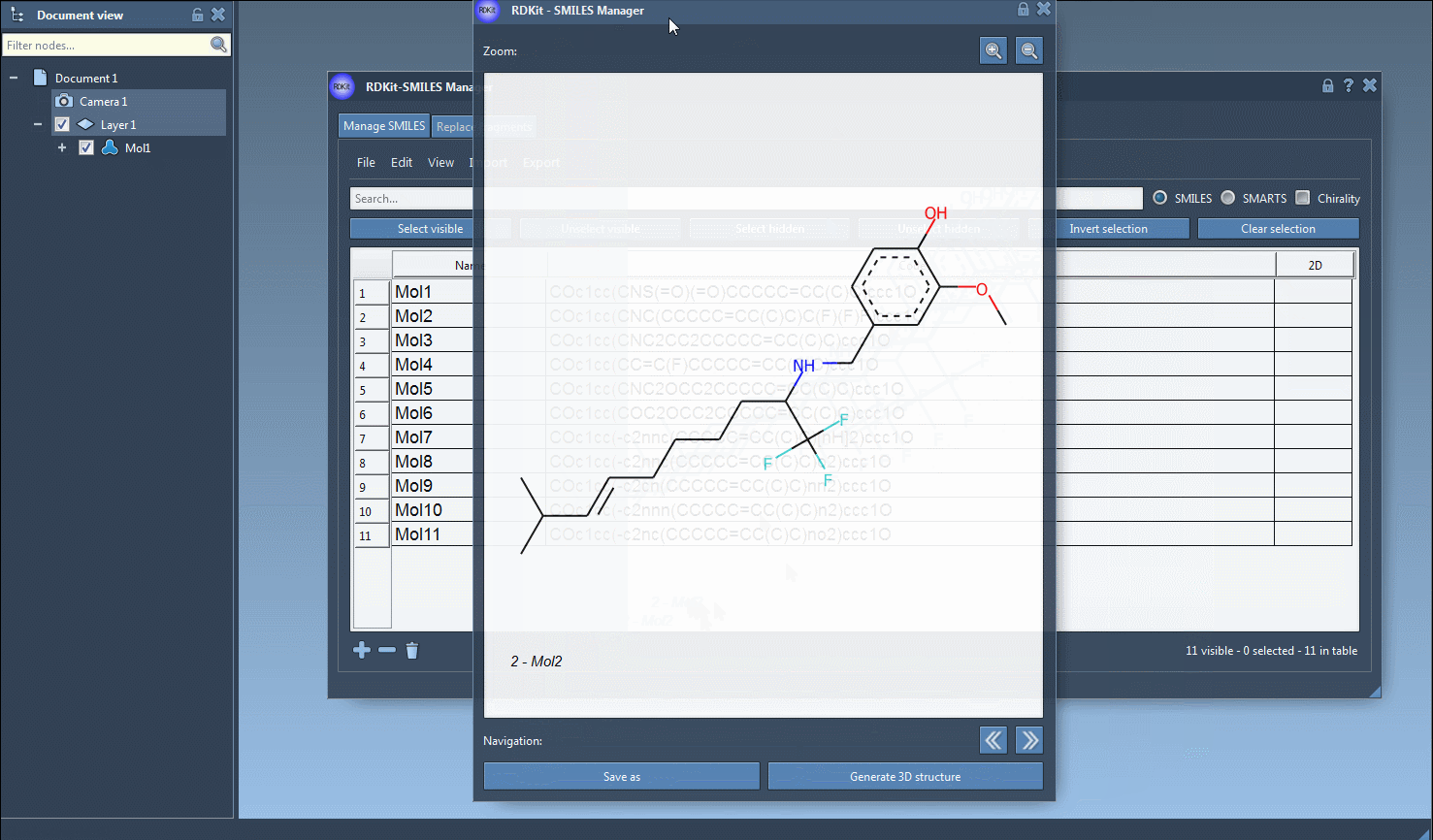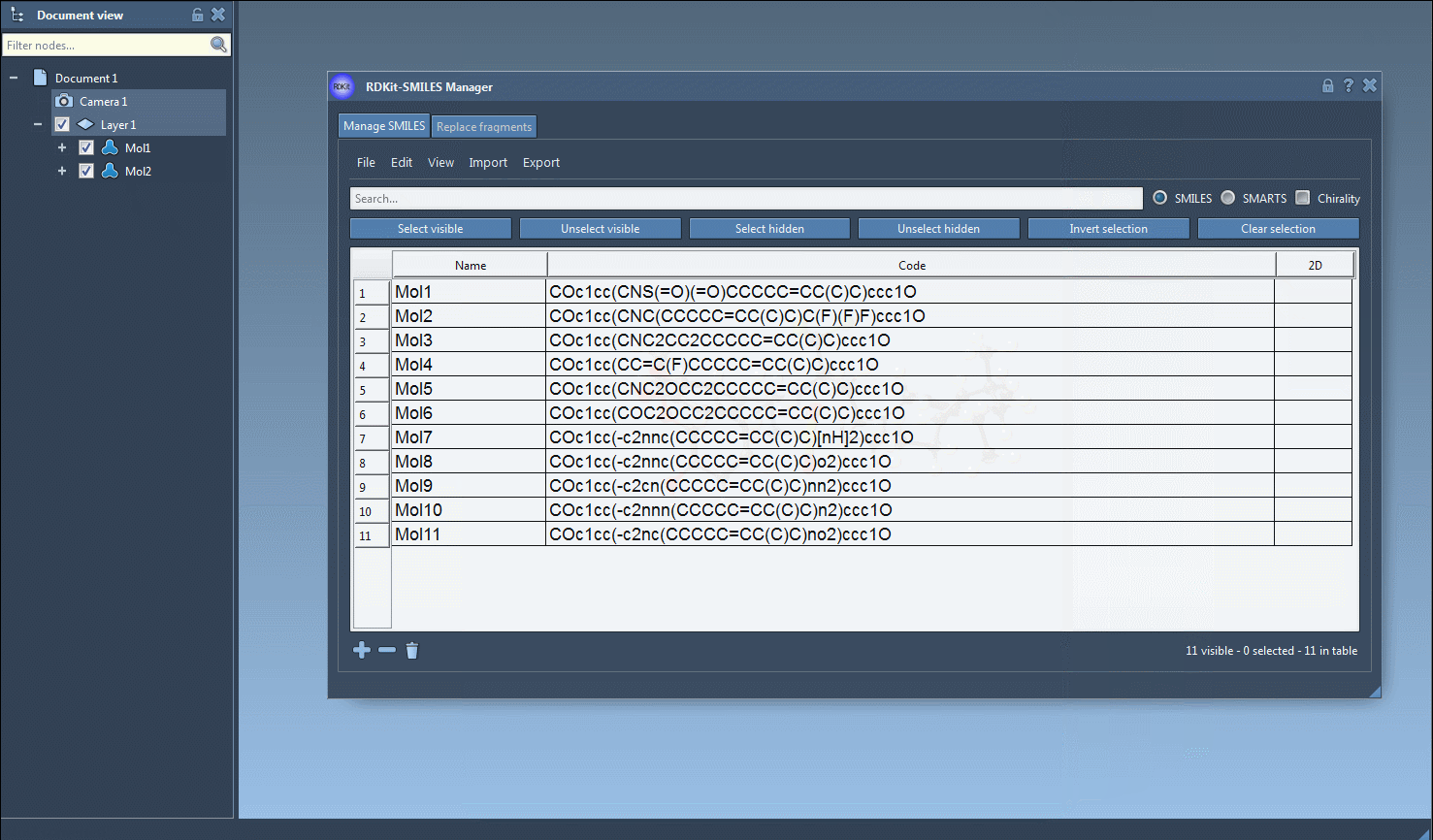
Manage SMILES strings#
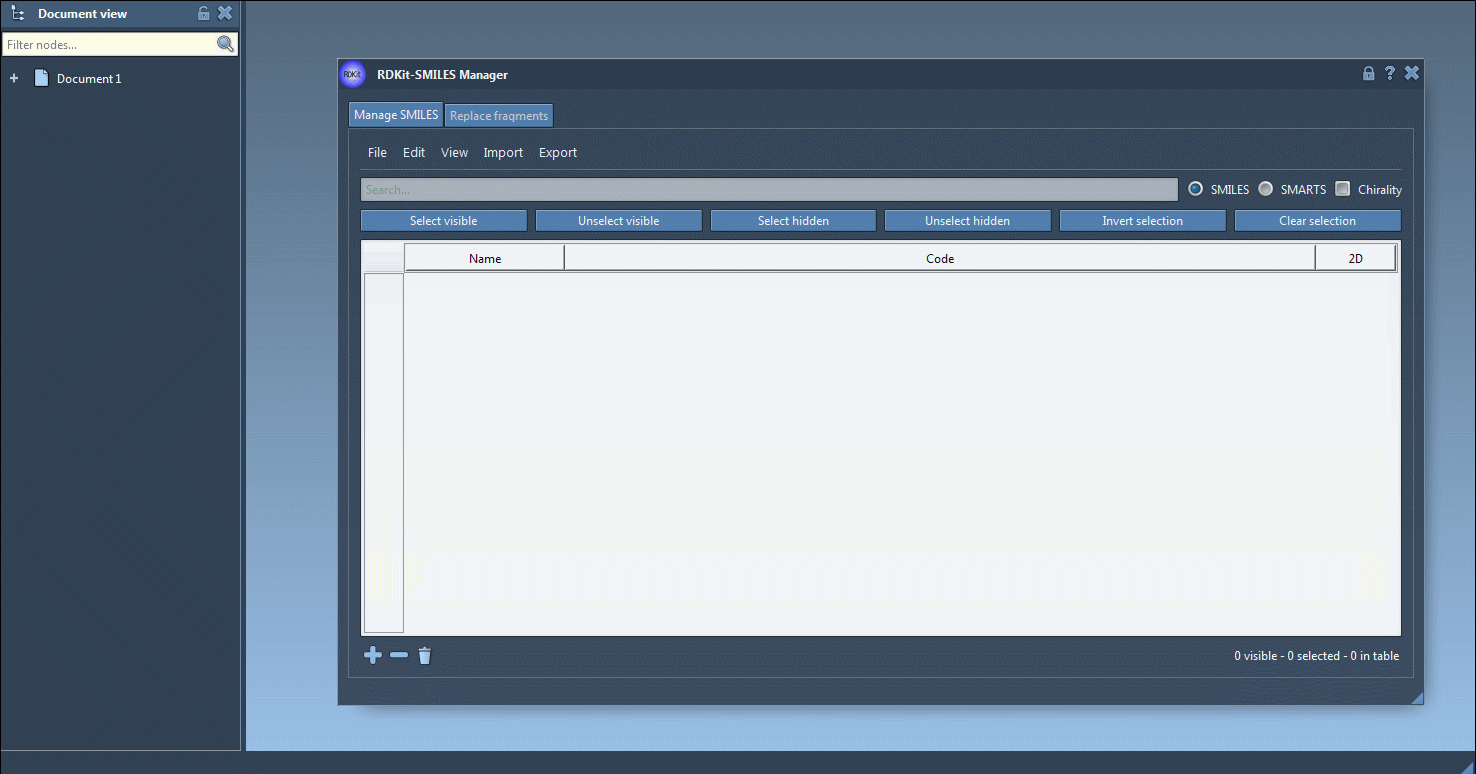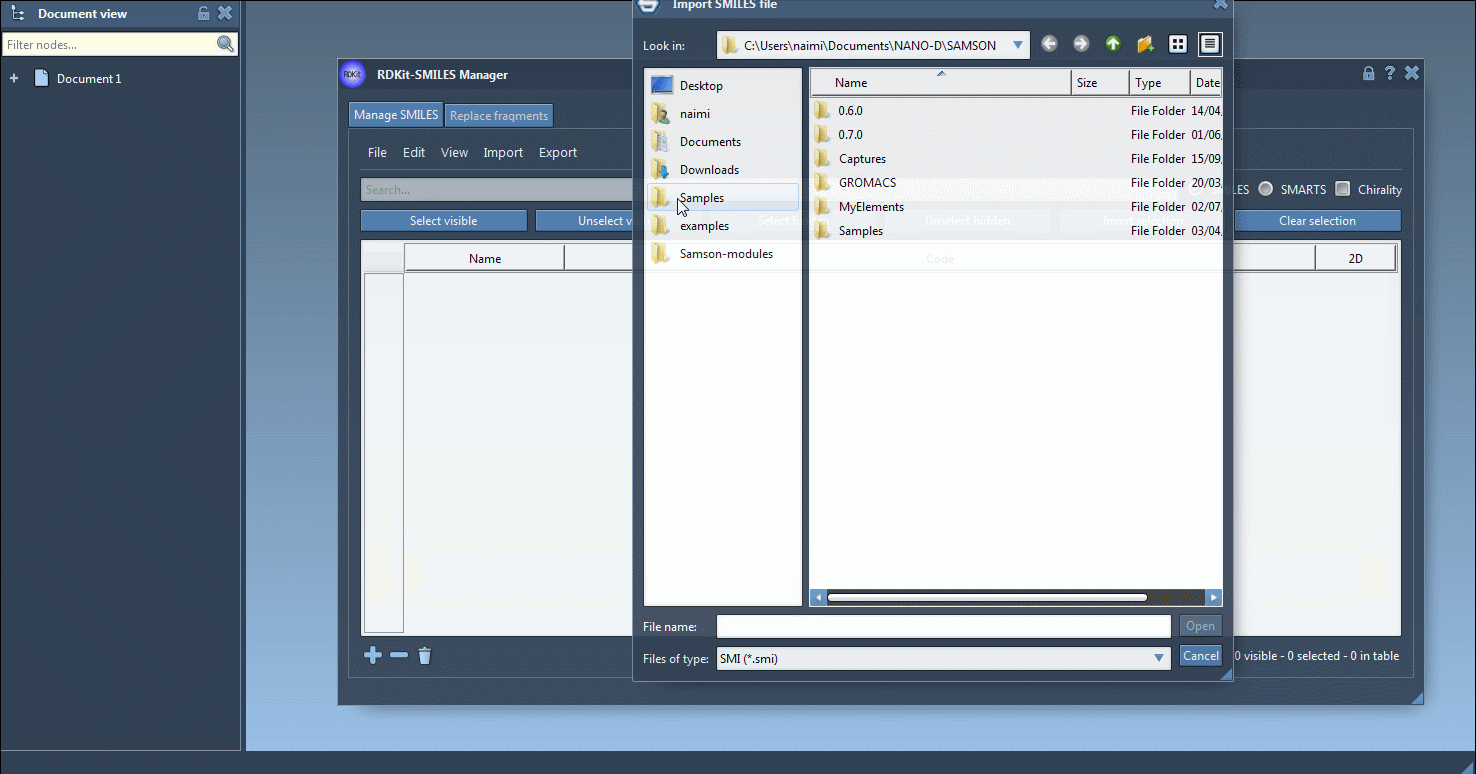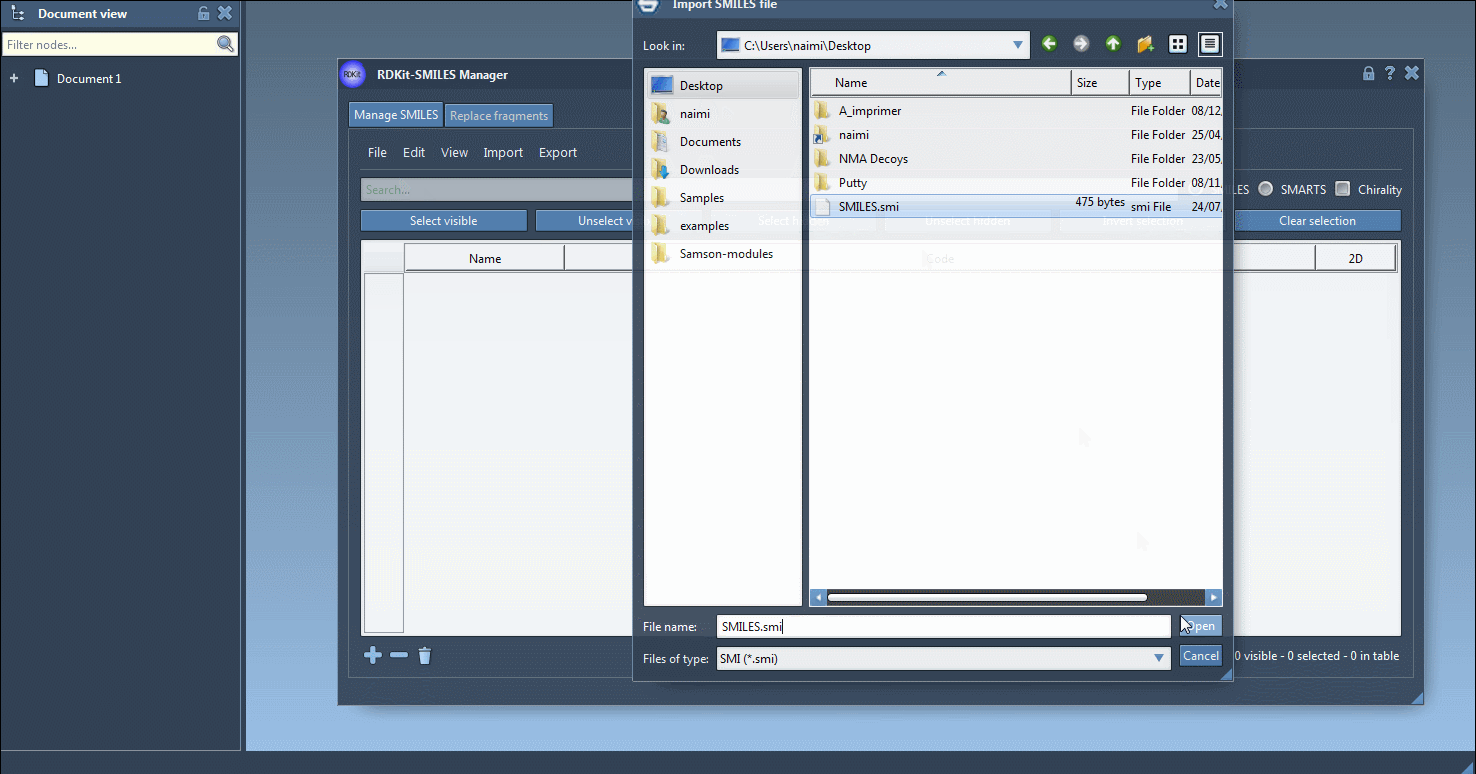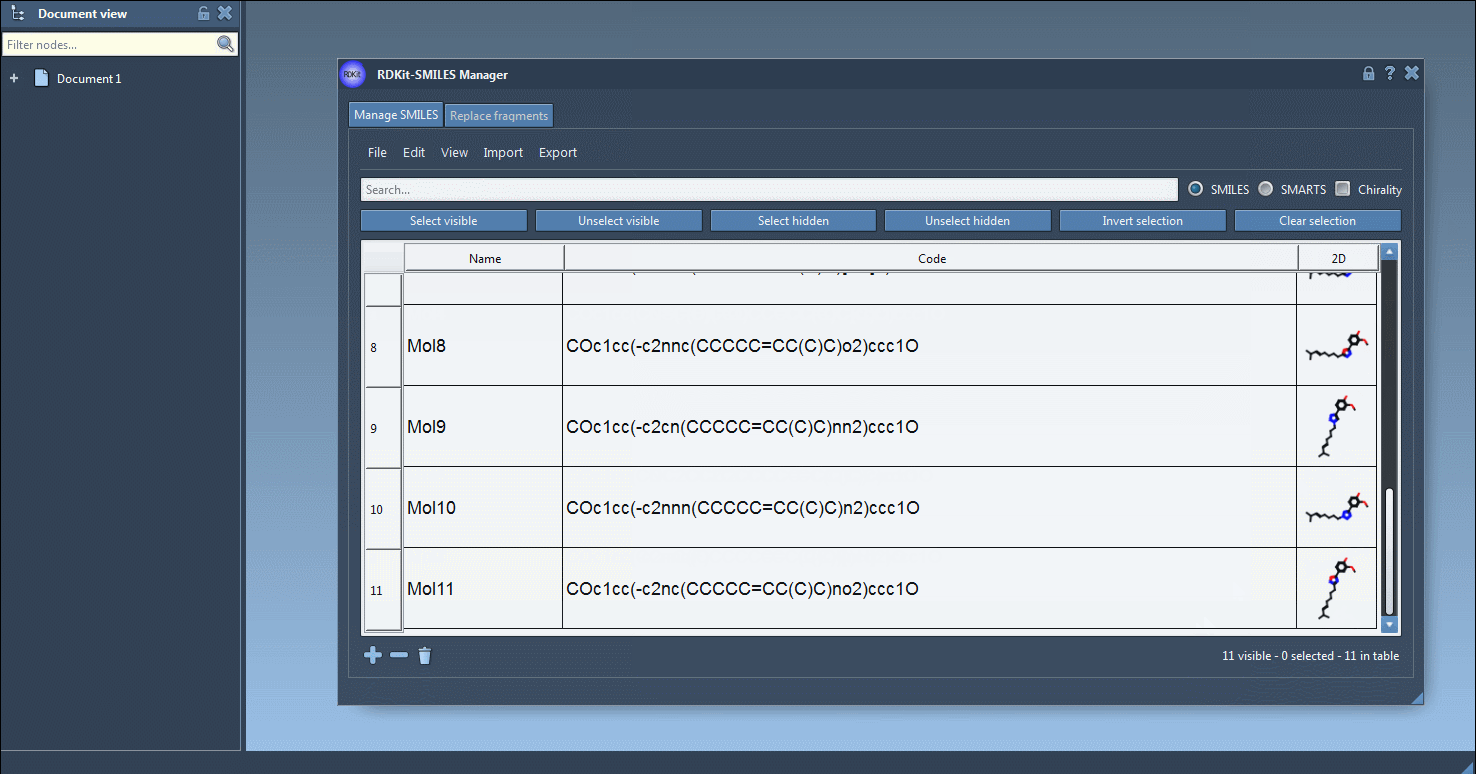
Import SMILES#
Import data by clicking Open in the File menu. The extension accepts the most common text-based SMILES inputs:
- RDKit-style
.smifiles with the SMILES string in the first column and the molecule name in the second column .txtfiles that contain one SMILES string per line- RDKit
.smifiles with extra attributes, which are ignored during import
The standard .smi layout is shown below.
SMILES Name
COc1cc(CNS(=O)(=O)CCCCC=CC(C)C)ccc1O Mol1
COc1cc(CNC(CCCCC=CC(C)C)C(F)(F)F)ccc1O Mol2
COc1cc(CNC2CC2CCCCC=CC(C)C)ccc1O Mol3
COc1cc(CC=C(F)CCCCC=CC(C)C)ccc1O Mol4
COc1cc(CNC2OCC2CCCCC=CC(C)C)ccc1O Mol5
COc1cc(COC2OCC2CCCCC=CC(C)C)ccc1O Mol6
If your RDKit SMILES file contains additional attributes, the extension still imports it, but only the SMILES code and molecule identifier are used.
SMILES Attribute Name CC 2 Mol1 C=C 5 Mol2 [CH+2]C[CH+2] 100 Mol3
Plain-text .txt files containing only SMILES strings can also be imported.
COc1cc(CNC(=O)CCCC/C=C/C(C)C)ccc1O
COc1cc(CNS(=O)(=O)CCCCC=CC(C)C)ccc1O
COc1cc(CNC(CCCCC=CC(C)C)C(F)(F)F)ccc1O
COc1cc(CNC2CC2CCCCC=CC(C)C)ccc1O
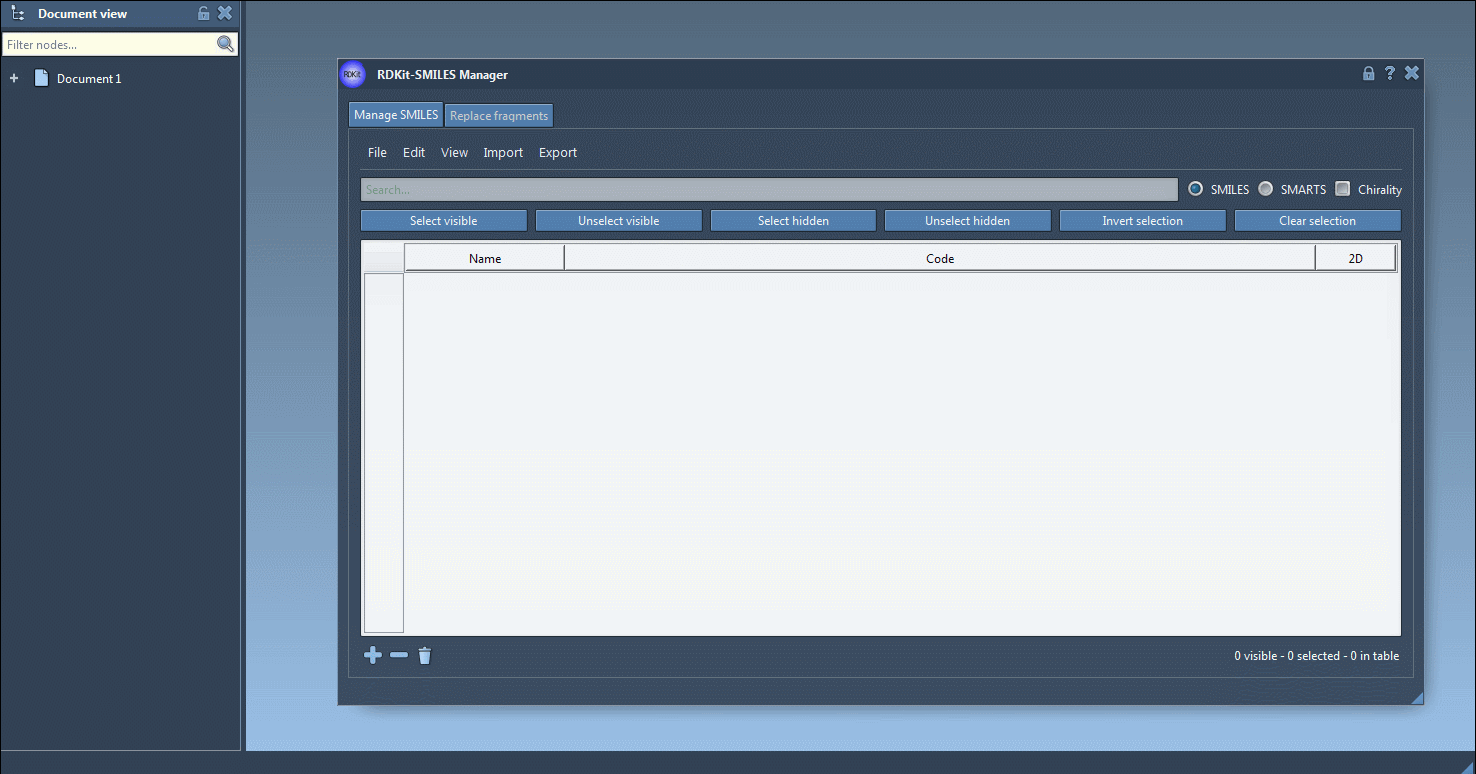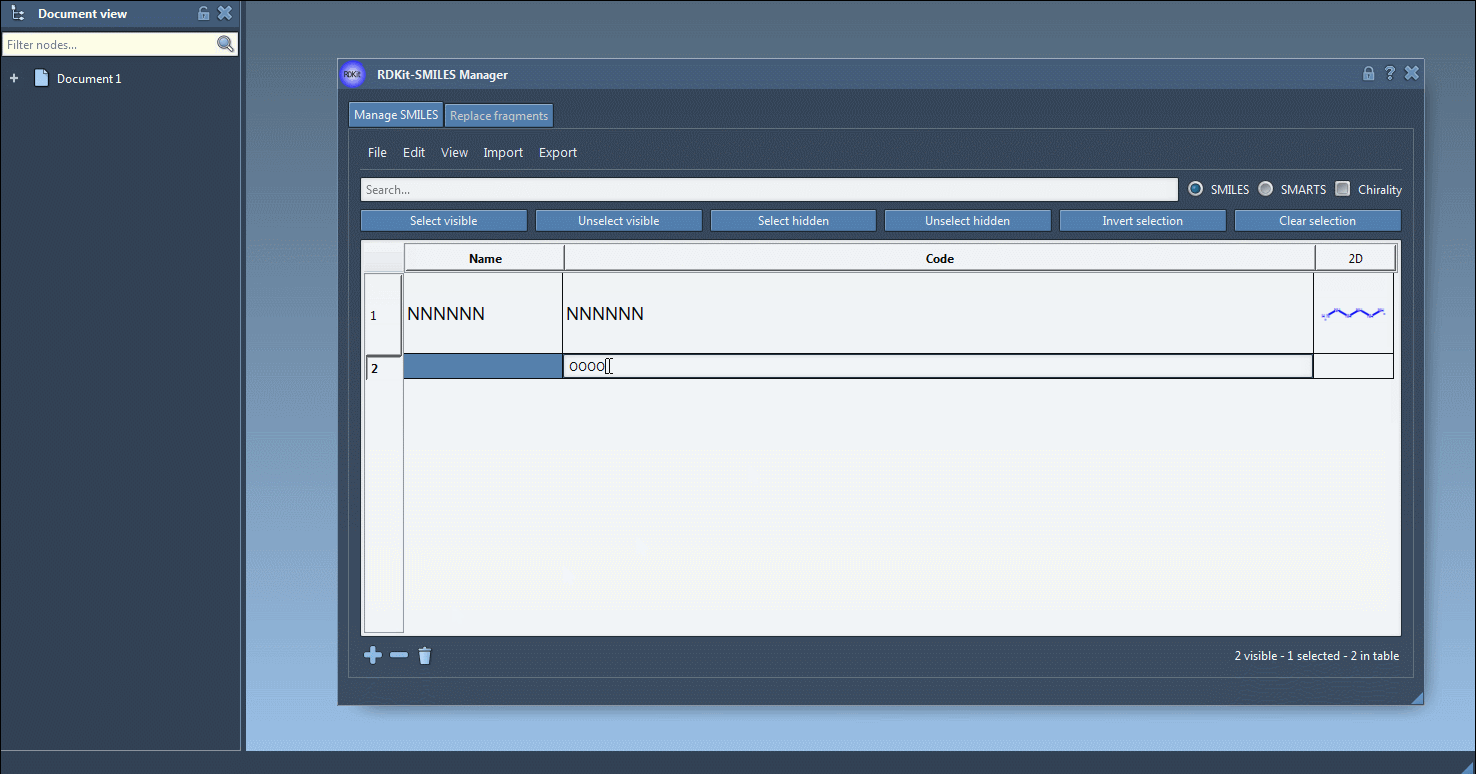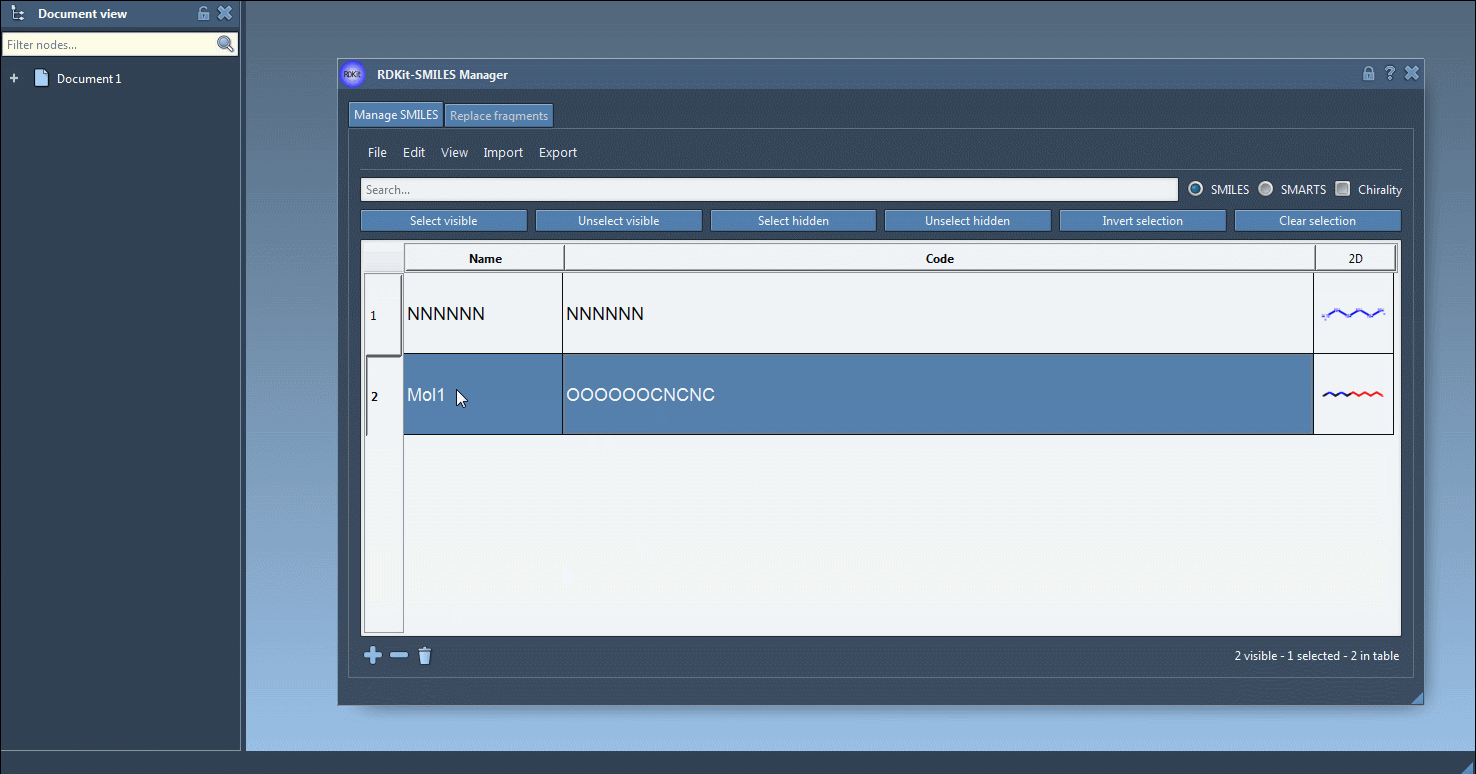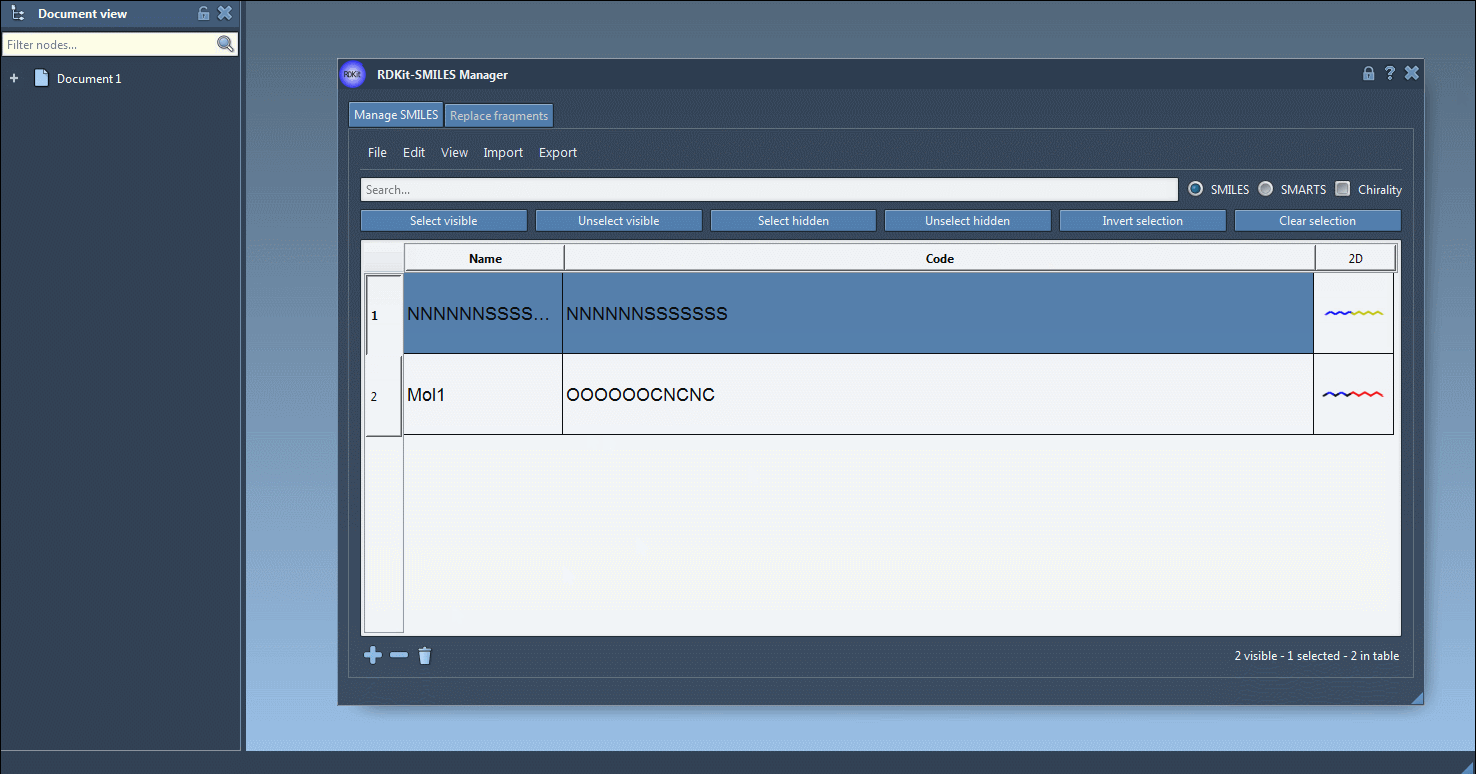
Adding, modifying, and removing SMILES#
You can manually add SMILES strings with the Add line ![]() action from the Edit menu or with the Add button
action from the Edit menu or with the Add button ![]() below the table. In the same way, you can remove individual entries or clear the table entirely. To edit an entry, modify the corresponding Code cell directly.
below the table. In the same way, you can remove individual entries or clear the table entirely. To edit an entry, modify the corresponding Code cell directly.
Also, a name can be assigned to each SMILES string. If no name has been assigned, the name will be the same as its SMILES code.
Generating, opening, updating, and saving 2D depictions#
Using RDKit functions, 2D depictions of the SMILES strings are generated on the fly. When a SMILES string is invalid, the corresponding line is highlighted and an error image is attributed instead of the 2D depiction. You can open a large view of the 2D depiction by either double-clicking on the 2D depiction or by right-clicking on the image and choosing the Open action in the context menu.

At any time, the 2D depictions presented in the table can be hidden by clicking on the Hide 2D image action of the View drop-down menu or by clicking on the 2D header cell of the table. Note that even when 2D depictions are hidden it is possible to open them using the aforementioned ways.
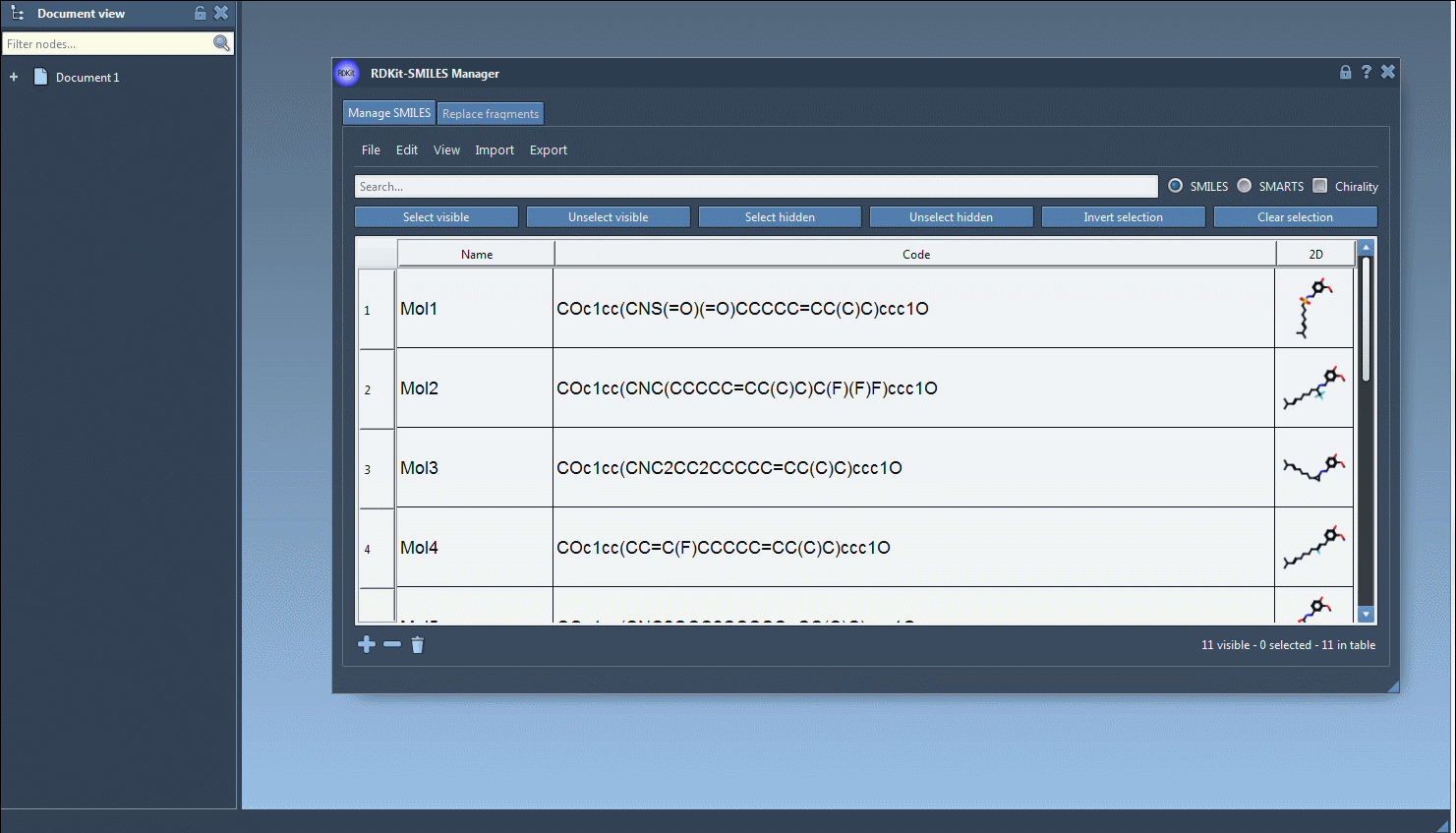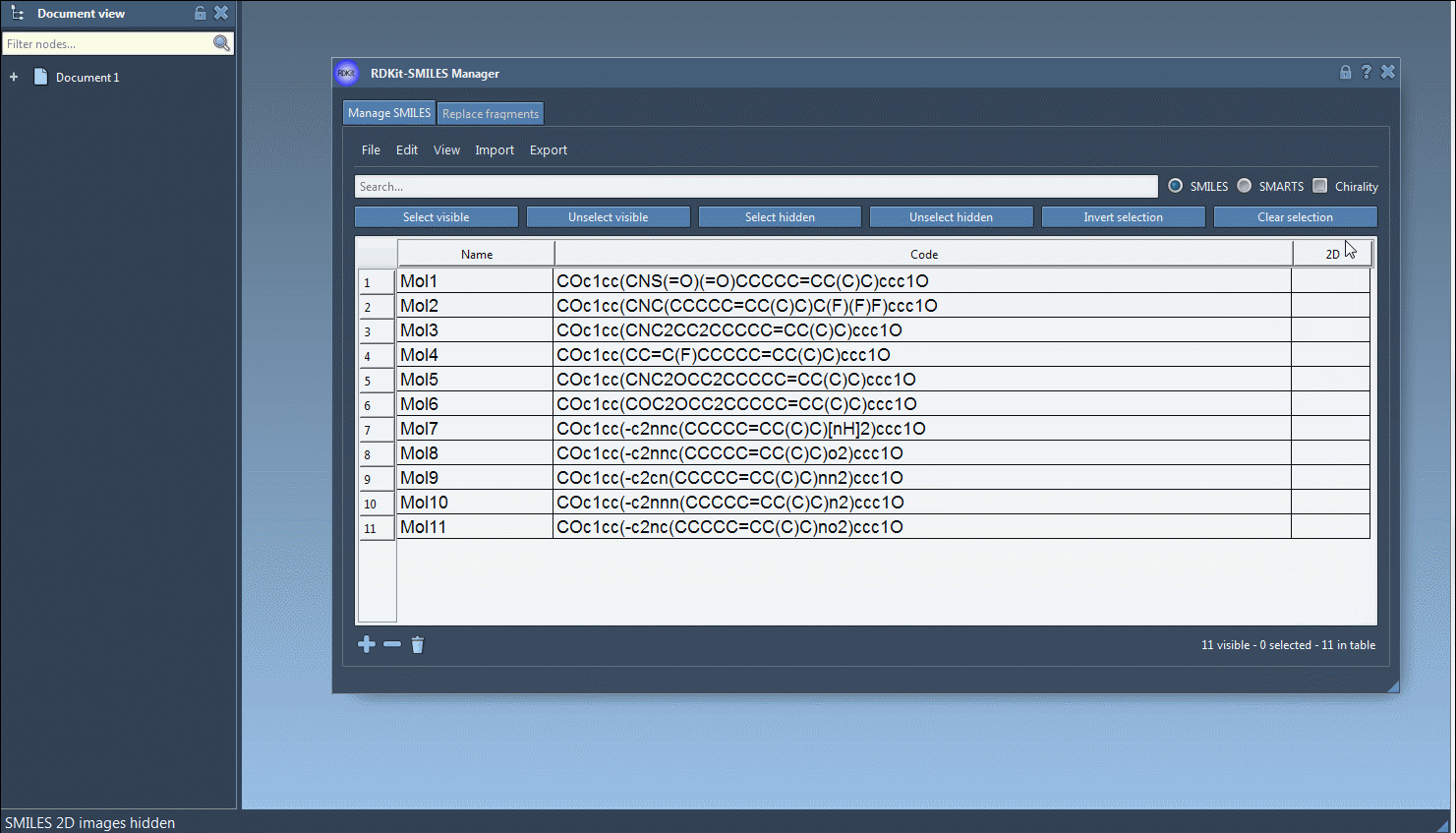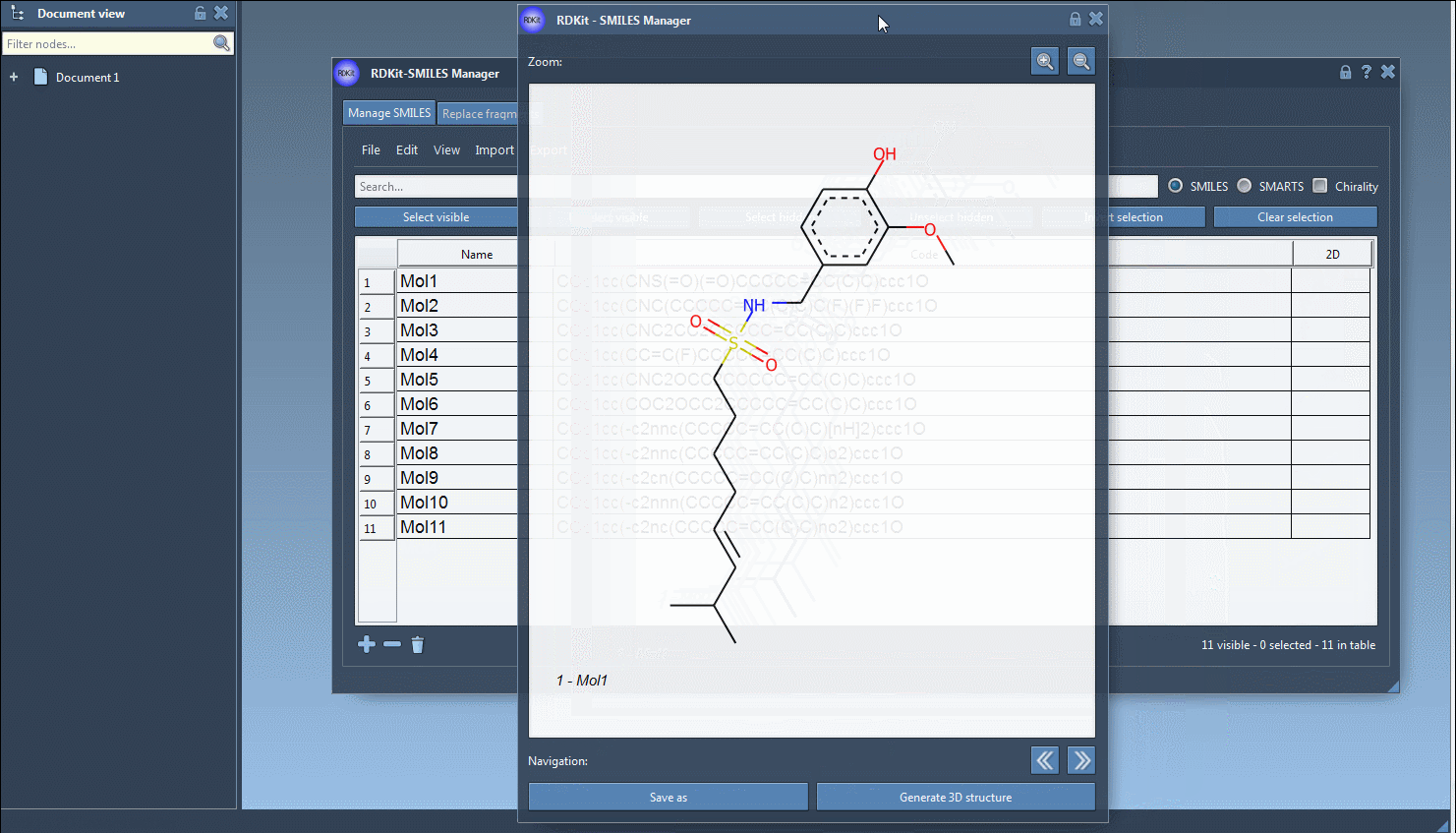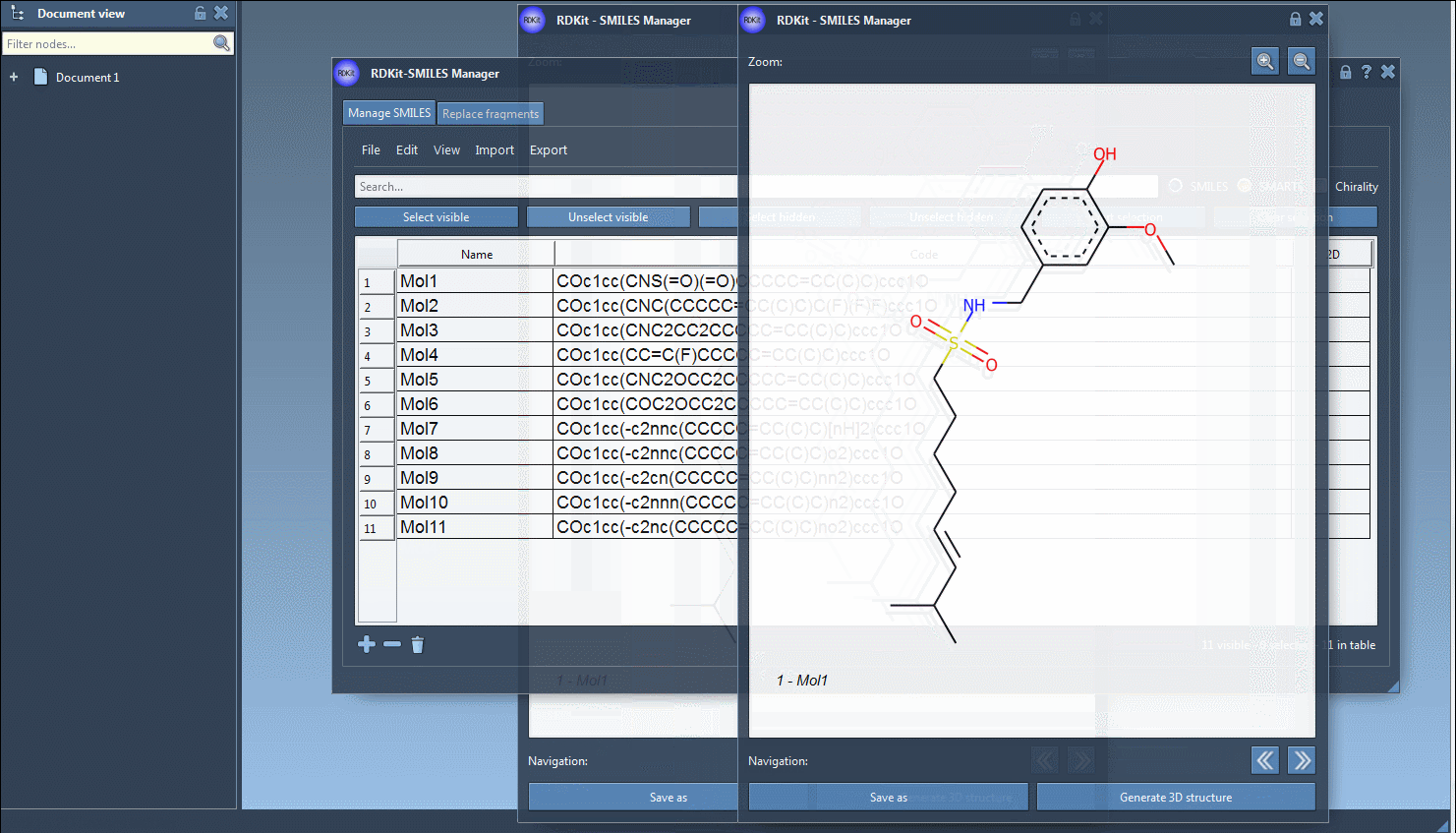
You can create several open windows to compare several molecules or navigate through all of them in a single window using the navigation buttons. For large molecules, it is possible to zoom in or out of the images. Note that when the SMILES code of a molecule is modified the corresponding image is automatically updated (the same for the molecule's name as well).
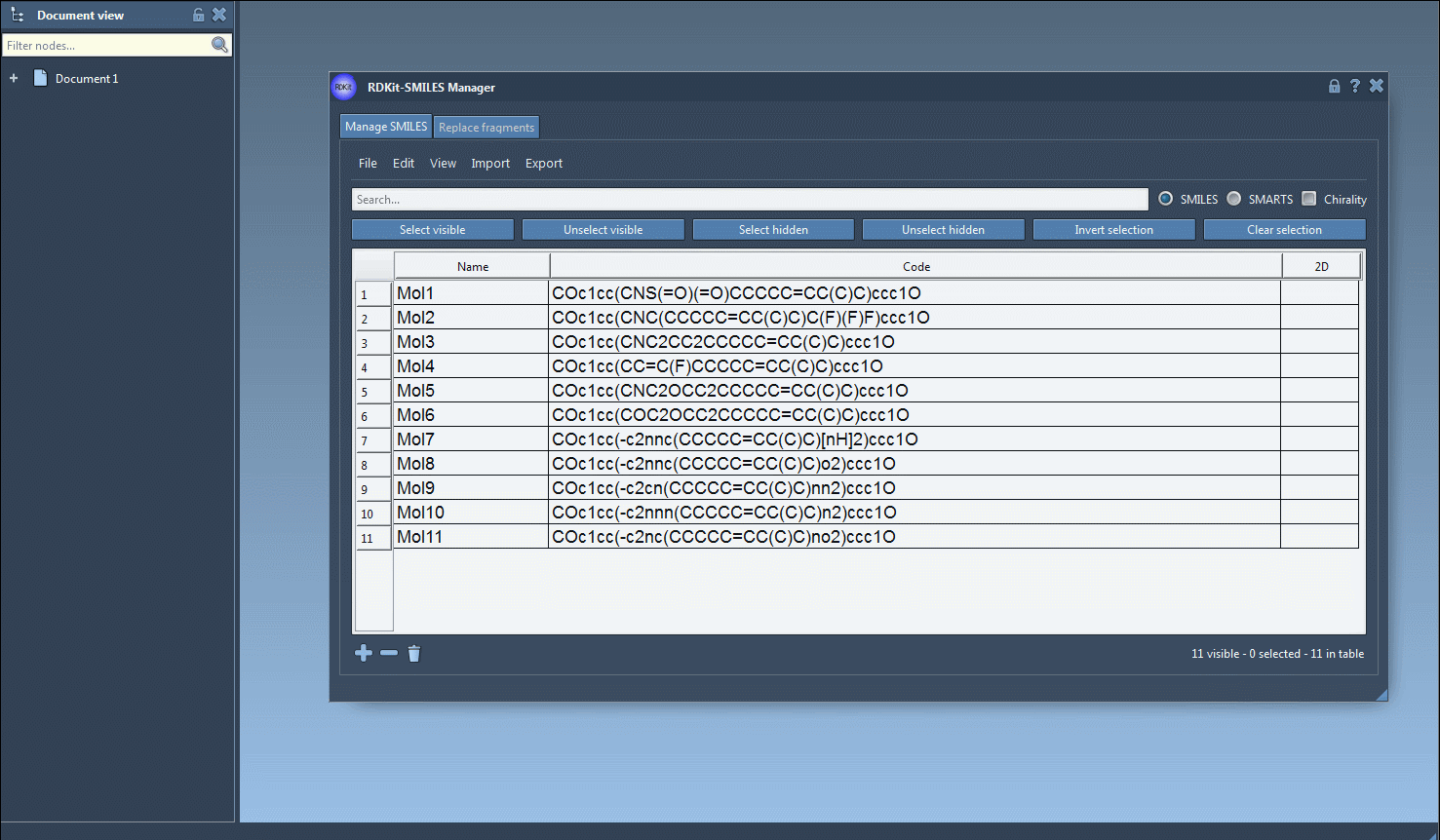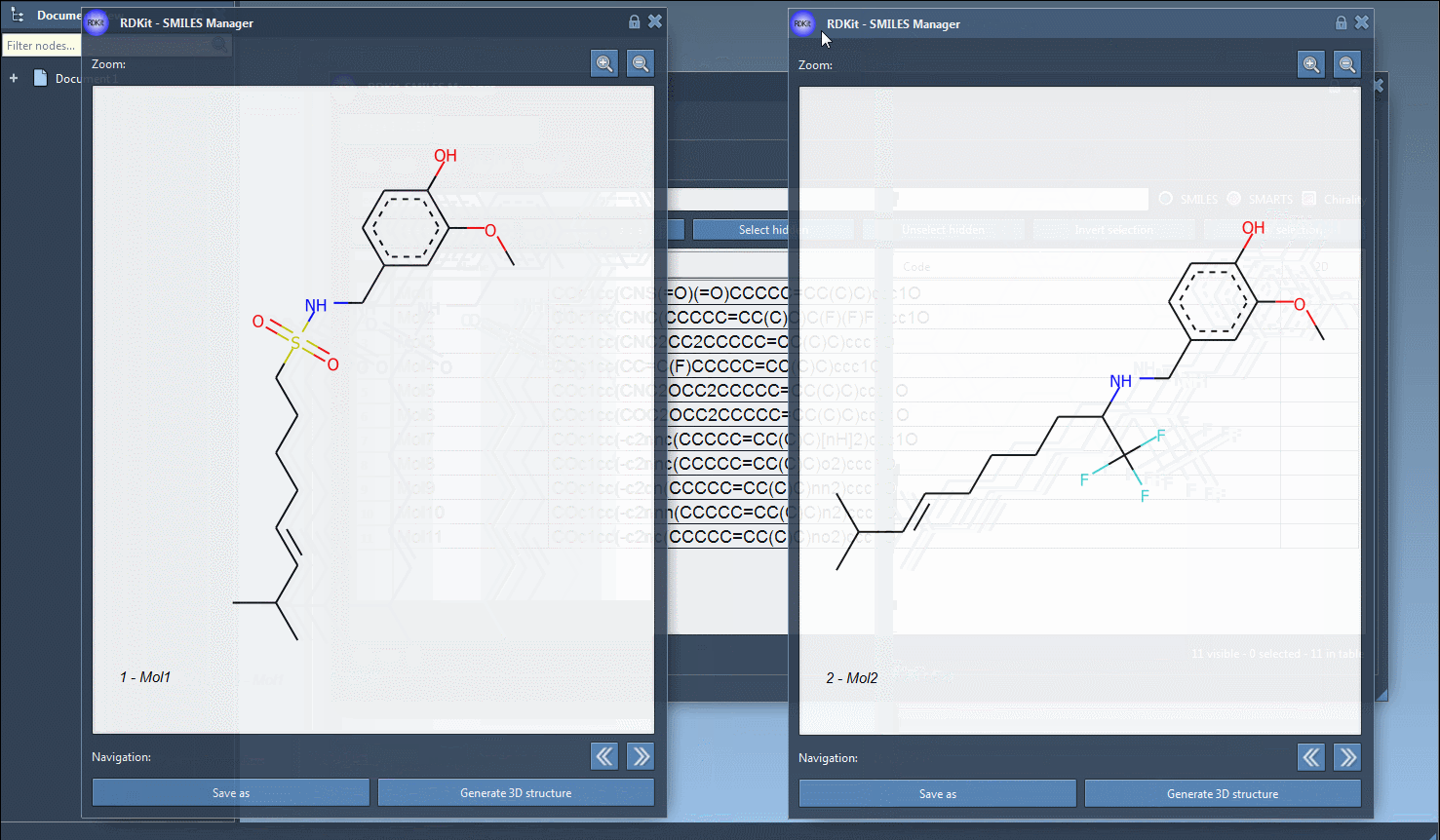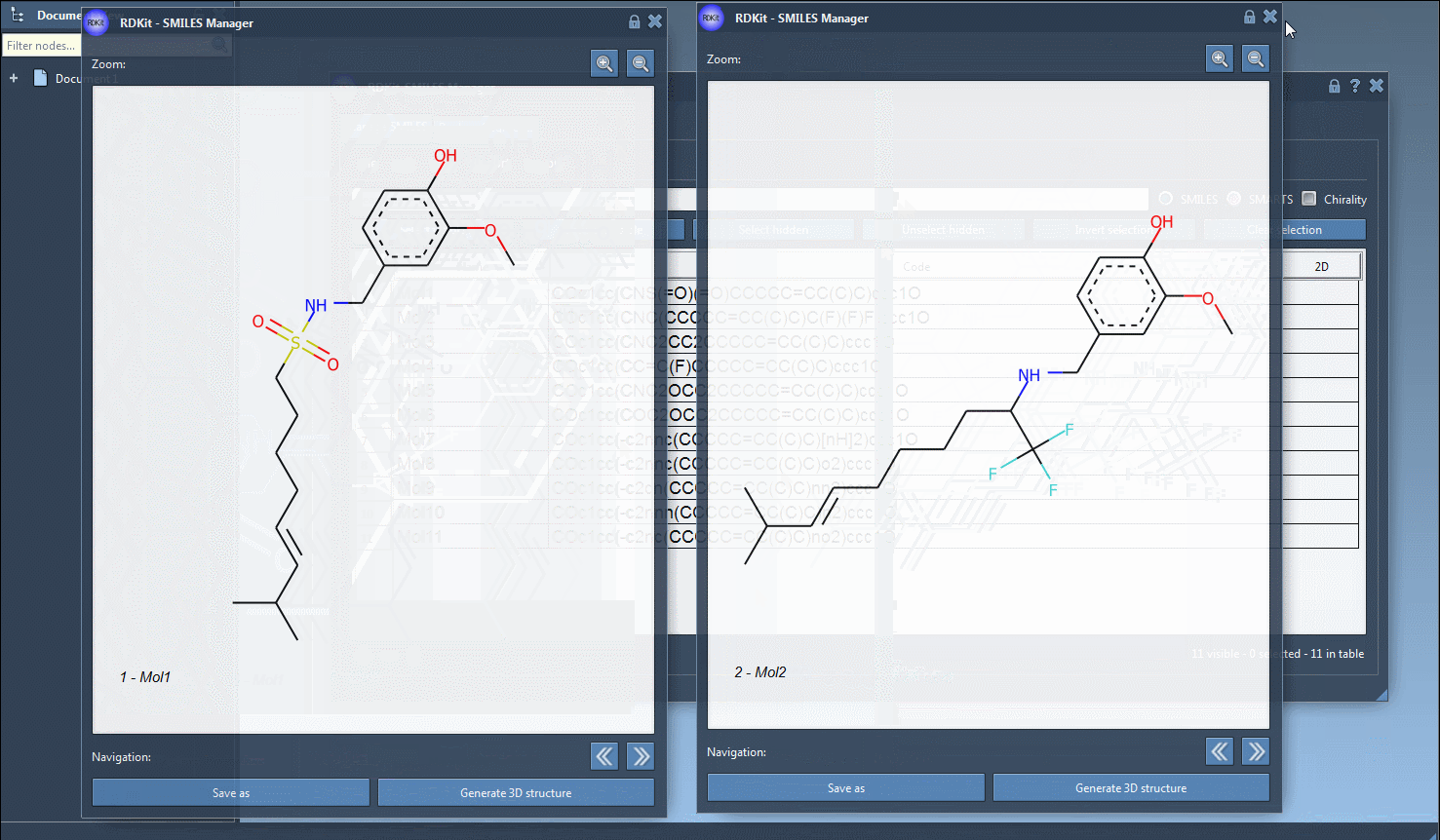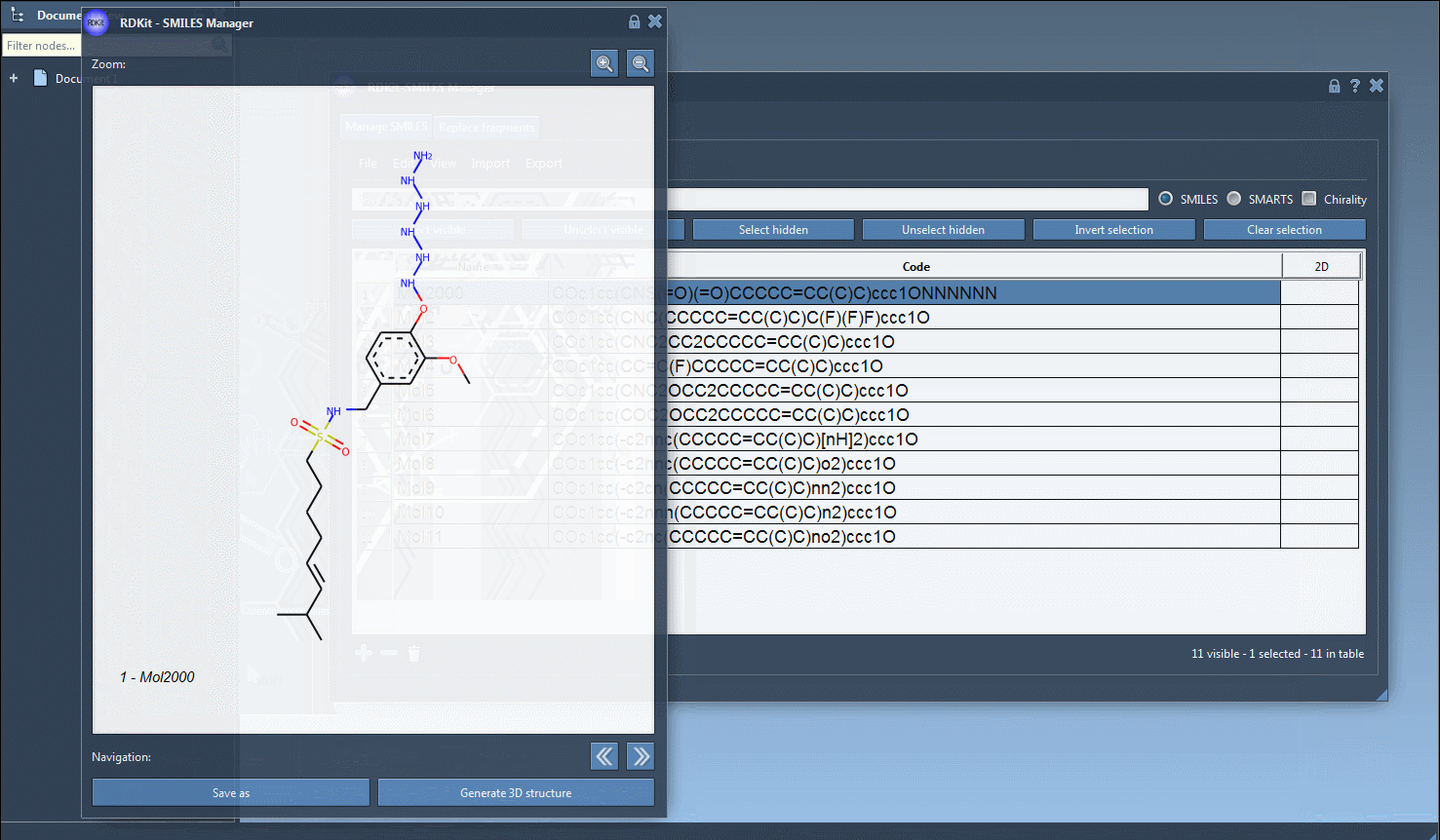
Finally, you can save the 2D depiction as a single PNG or SVG image either from the right-click context menu or from the large view window.
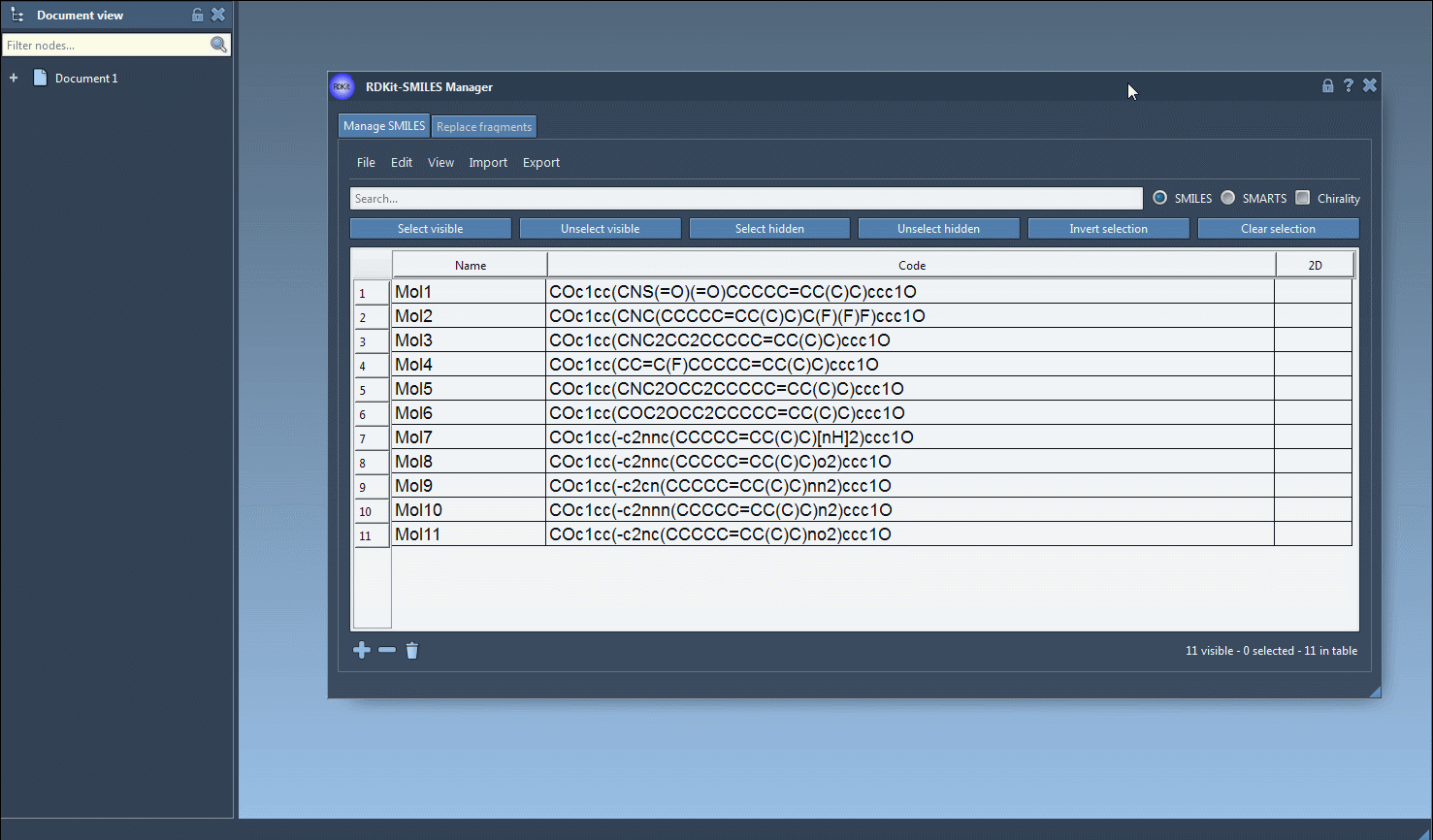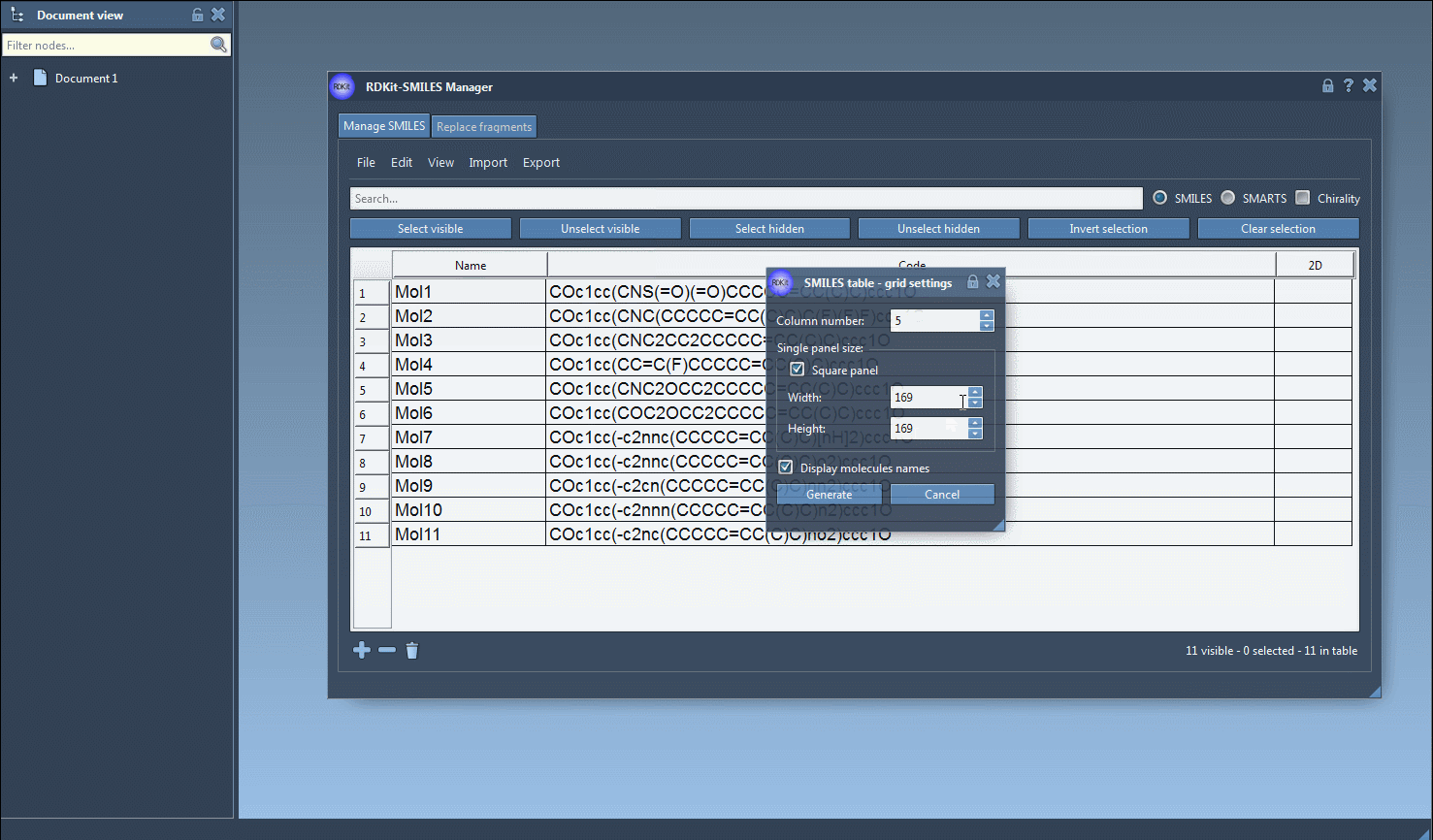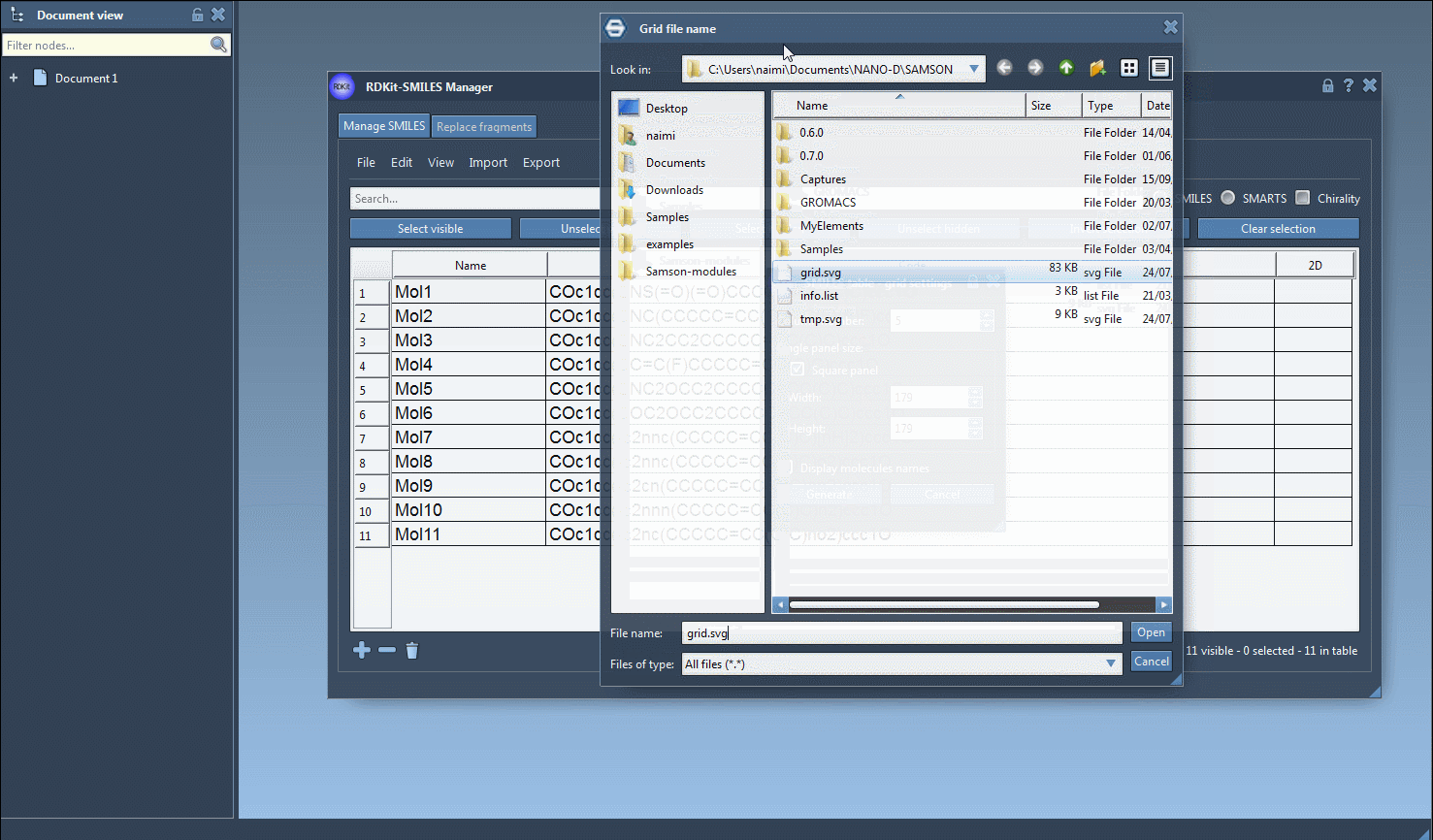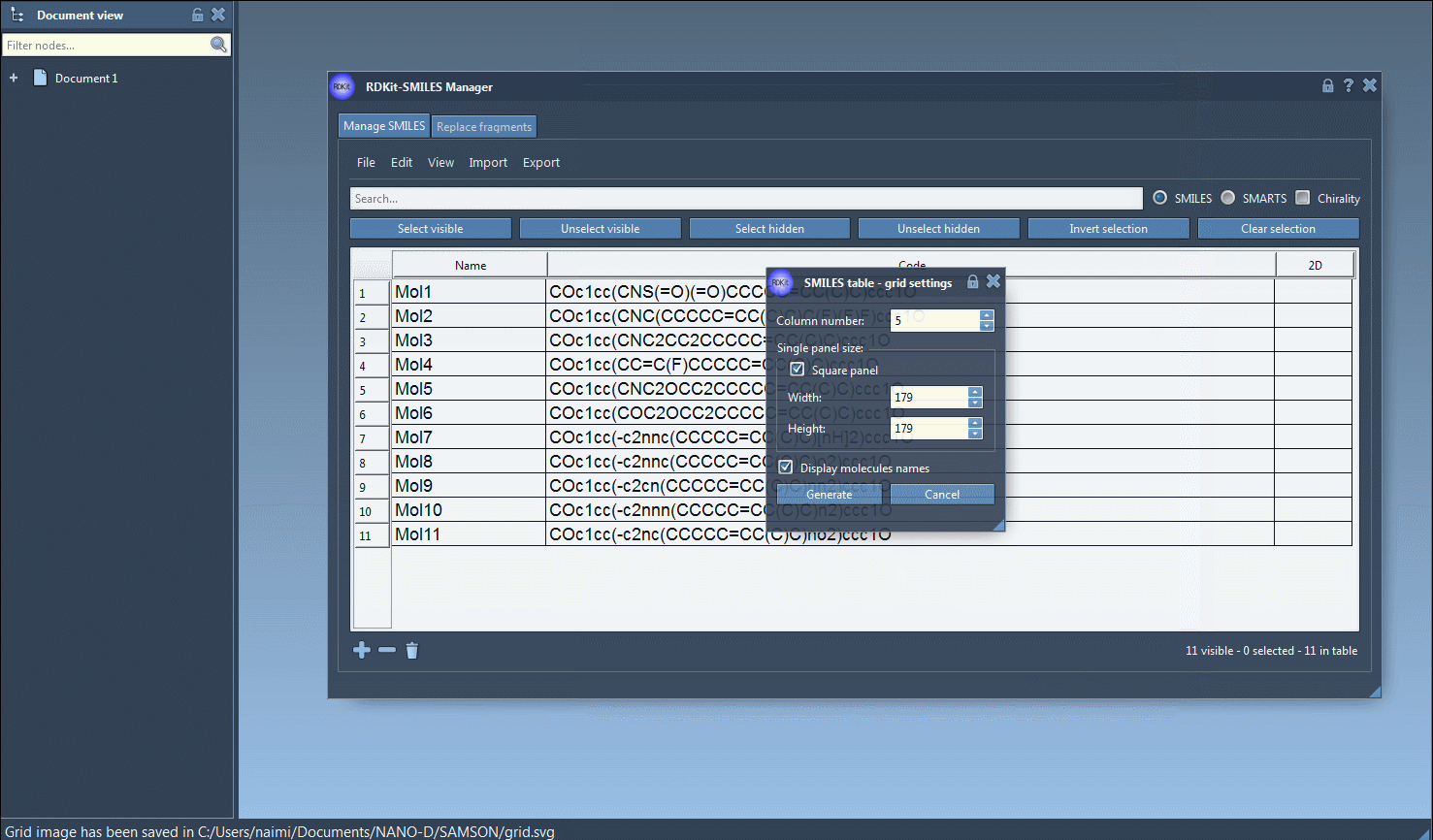
Grid images of 2D depictions#
It is also possible to save the 2D depictions in a grid in an SVG file by clicking on the Save as grid image action of the File drop-down menu. You can also save only the selected 2D depictions by choosing the Save selection as grid image action. A popup window will appear where you will be able to set the parameters of the grid (number of columns, size of a single panel, and whether the molecule's name should be displayed or not).
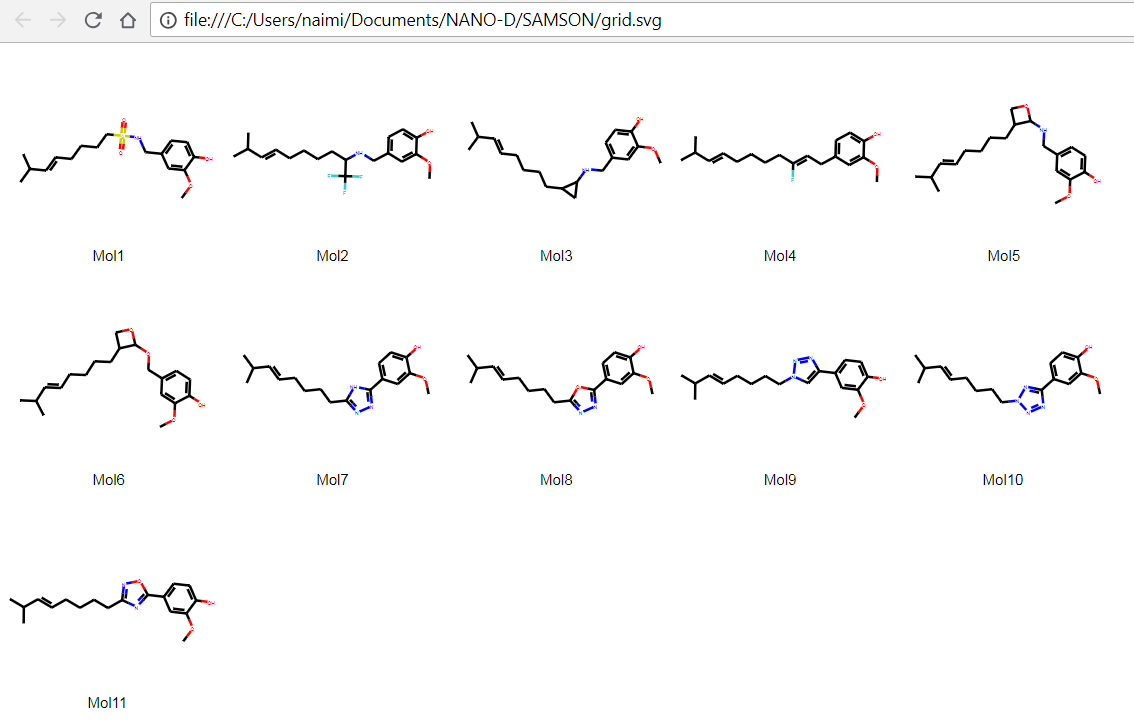
Generating 3D structures#
The main feature of this SAMSON module is to generate 3D structures from SMILES codes. After selecting molecules in the table, you can click on the Selected SMILES string to Document action in the Export drop-down menu. The 3D structures of the selected molecules with their corresponding names will be added to the active document.
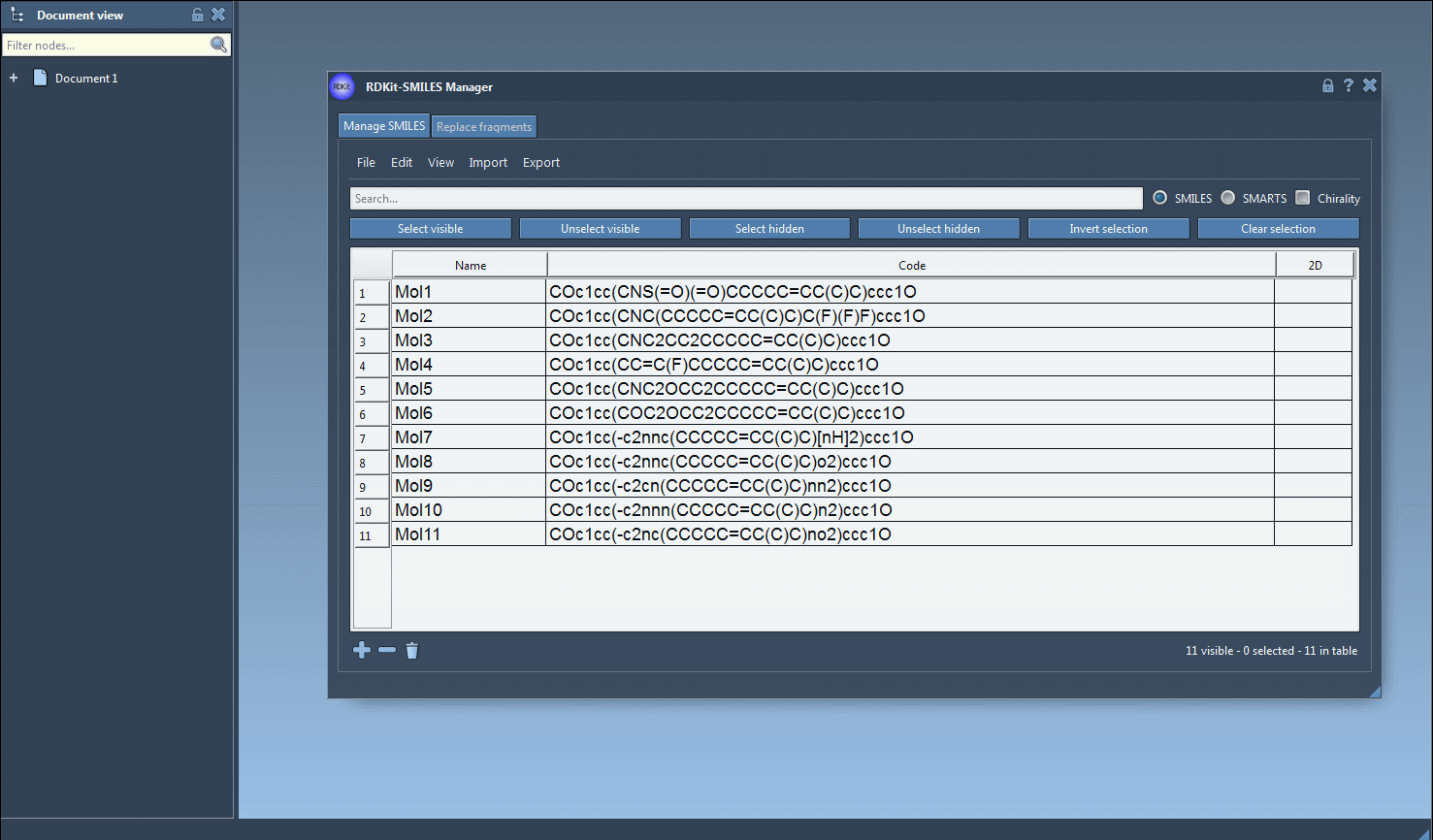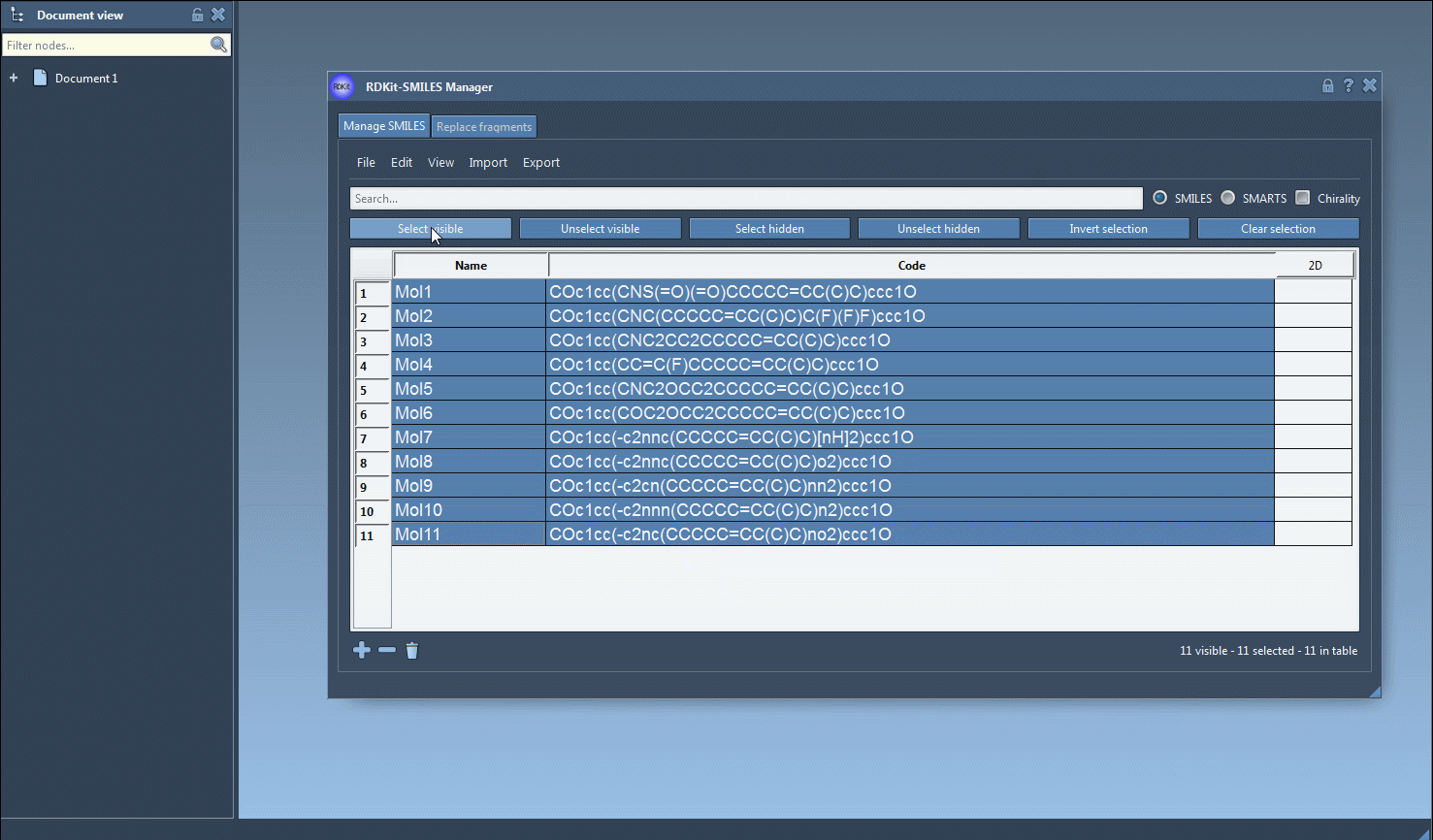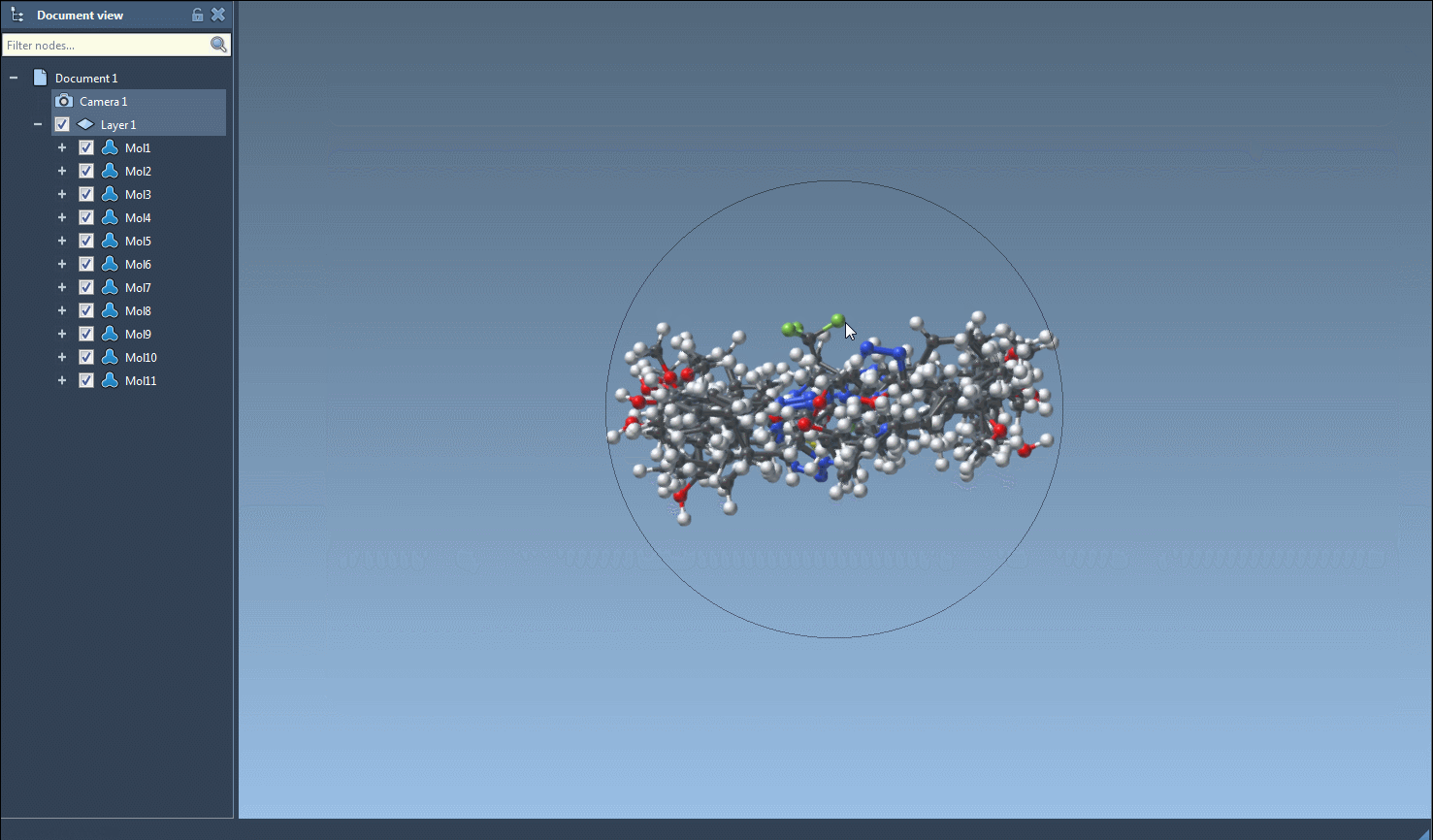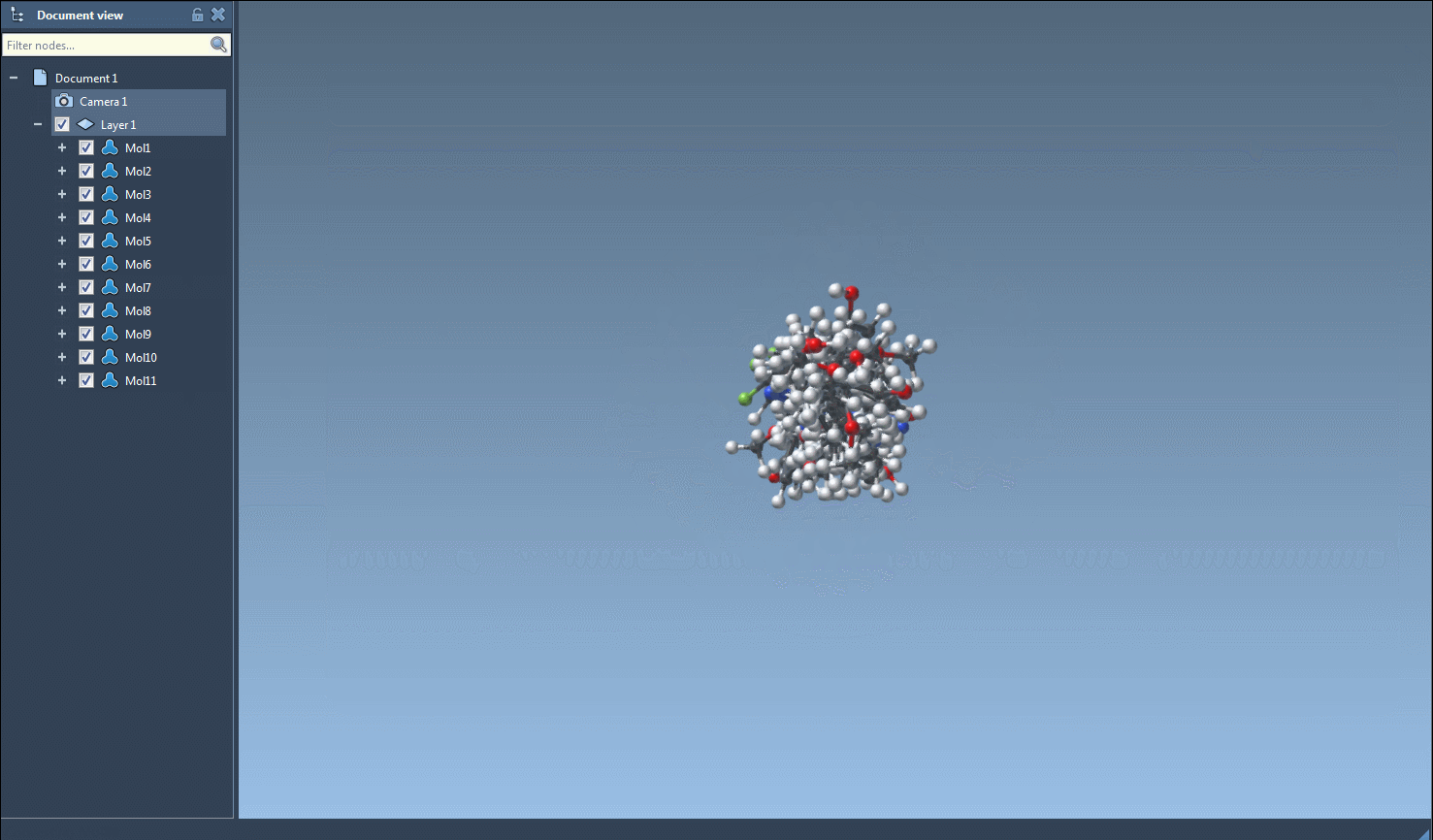
The generation of the 3D structure can also be done for a single SMILES string either by clicking on the Generate 3D structure action in the right-click context menu or by clicking on the Generate 3D structure button in the large view window.
Substructure search#
One interesting feature that I have improved in this new version is the substructure search. It is now possible to do a more advanced search using a given combination of patterns. For example, let us generate only the molecules that have a fluorine atom and a nitrogen atom separated in space. For that, in the Search bar enter F for fluorine then press Select visible. This will select SMILES strings having a fluorine atom (two SMILES strings in the animation below). Then we clear the search bar. Note that our selection remains unchanged (in the animation below, two SMILES strings are still selected). Now we enter N for nitrogen and we can see in the result of our search that only one selected SMILES string is present (the other one does not have nitrogen). So we press the Unselect hidden button in order to unselect the molecule that is not displayed and keep only the ones presenting the nitrogen atom.
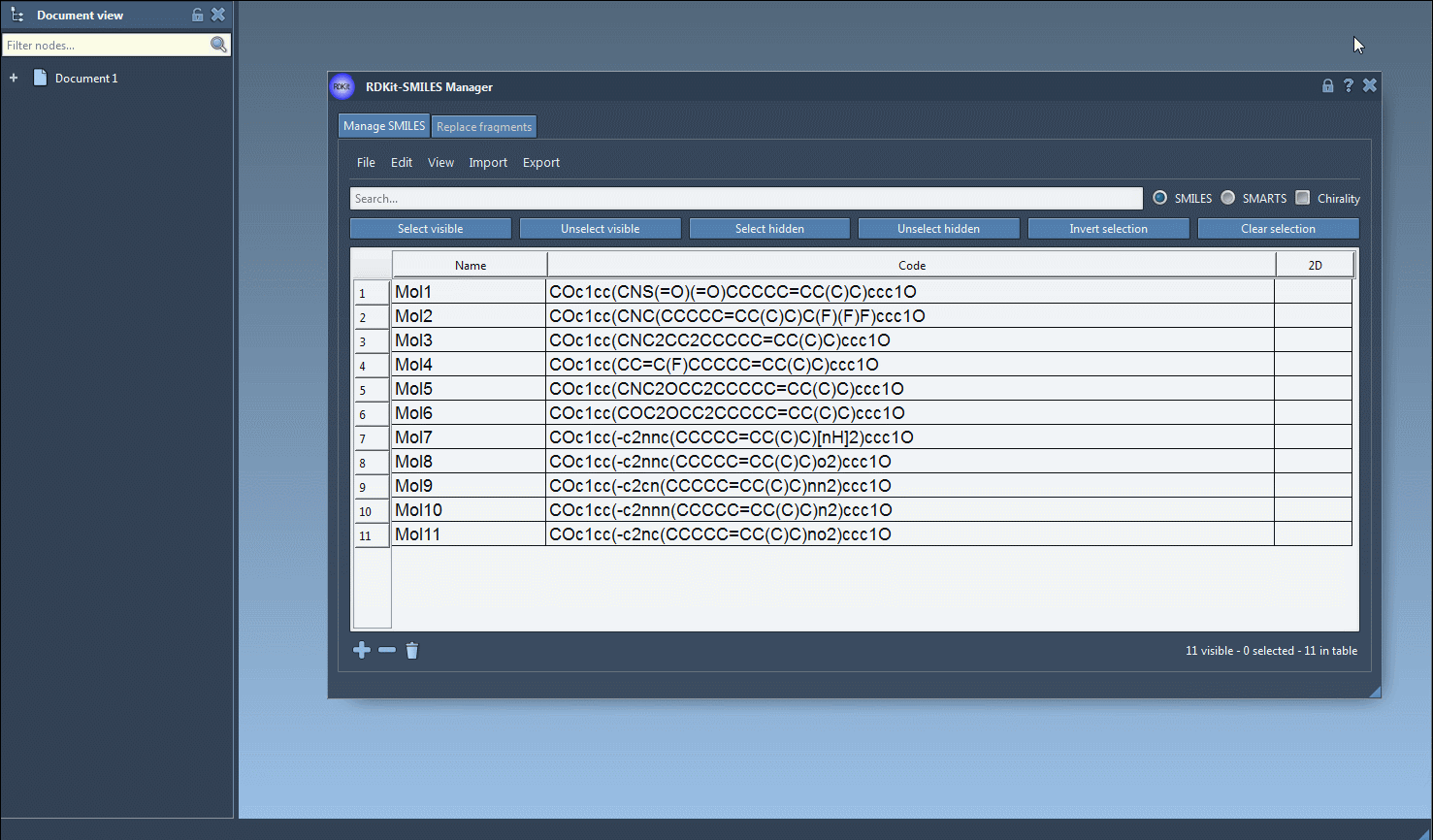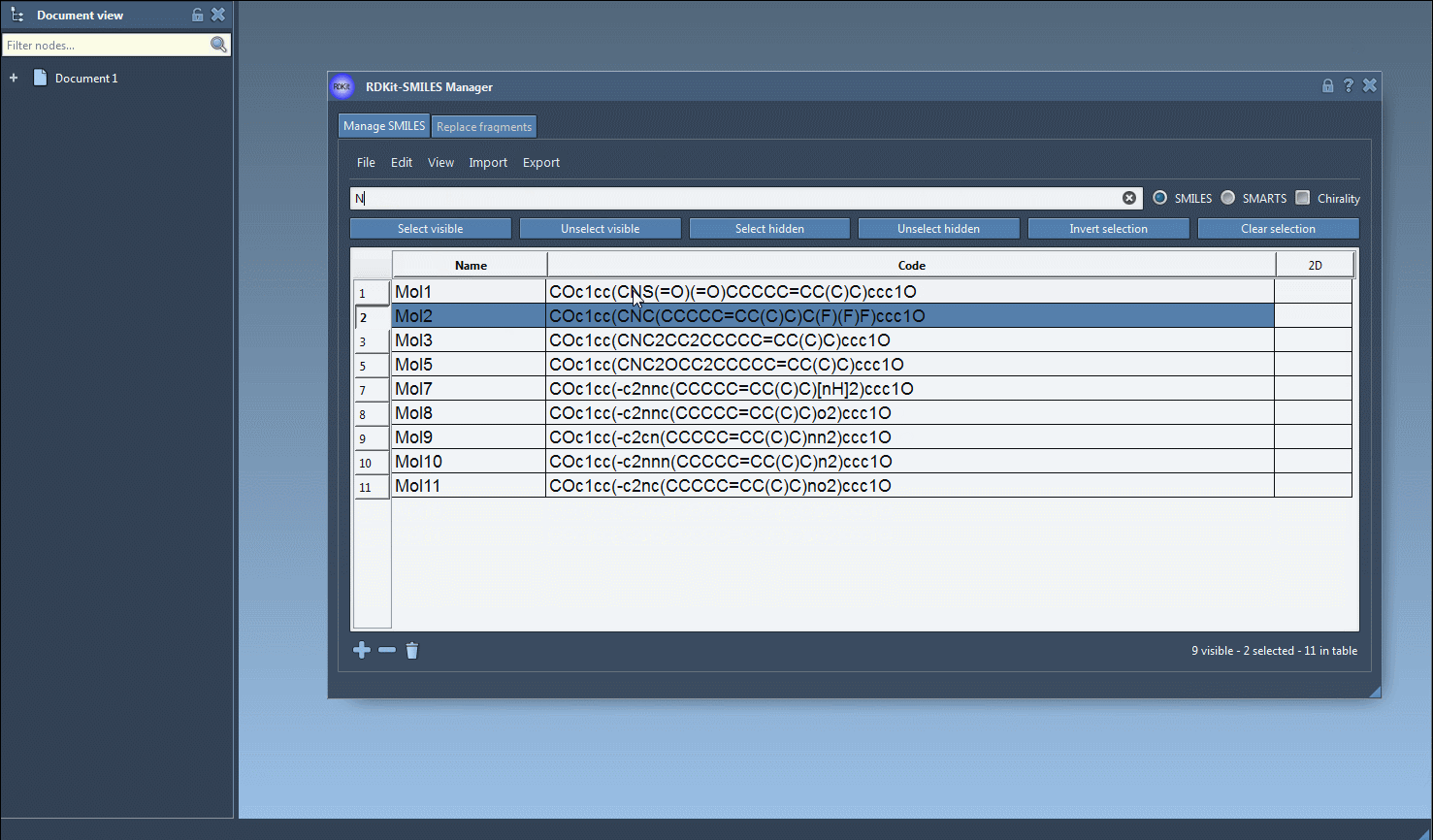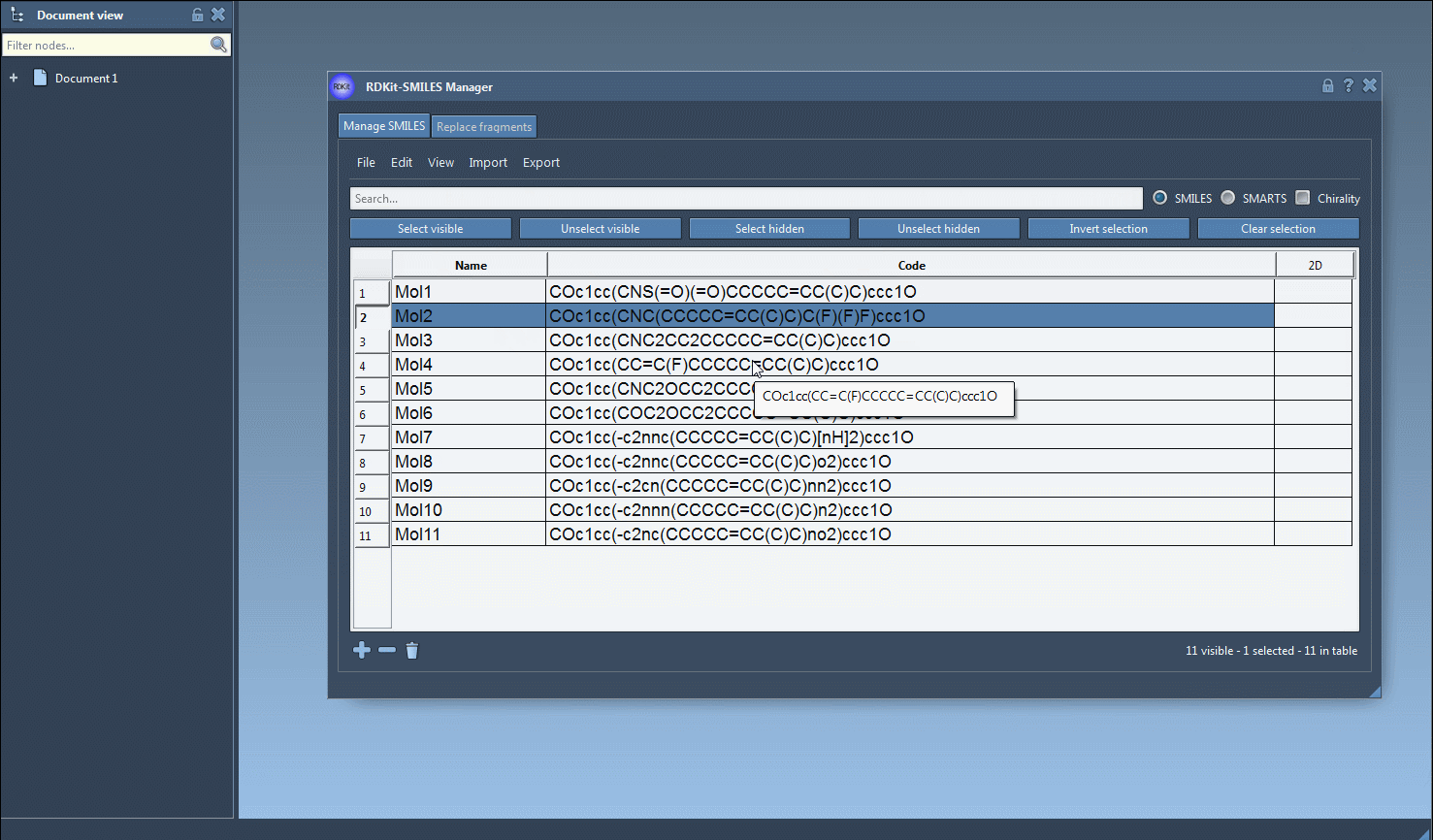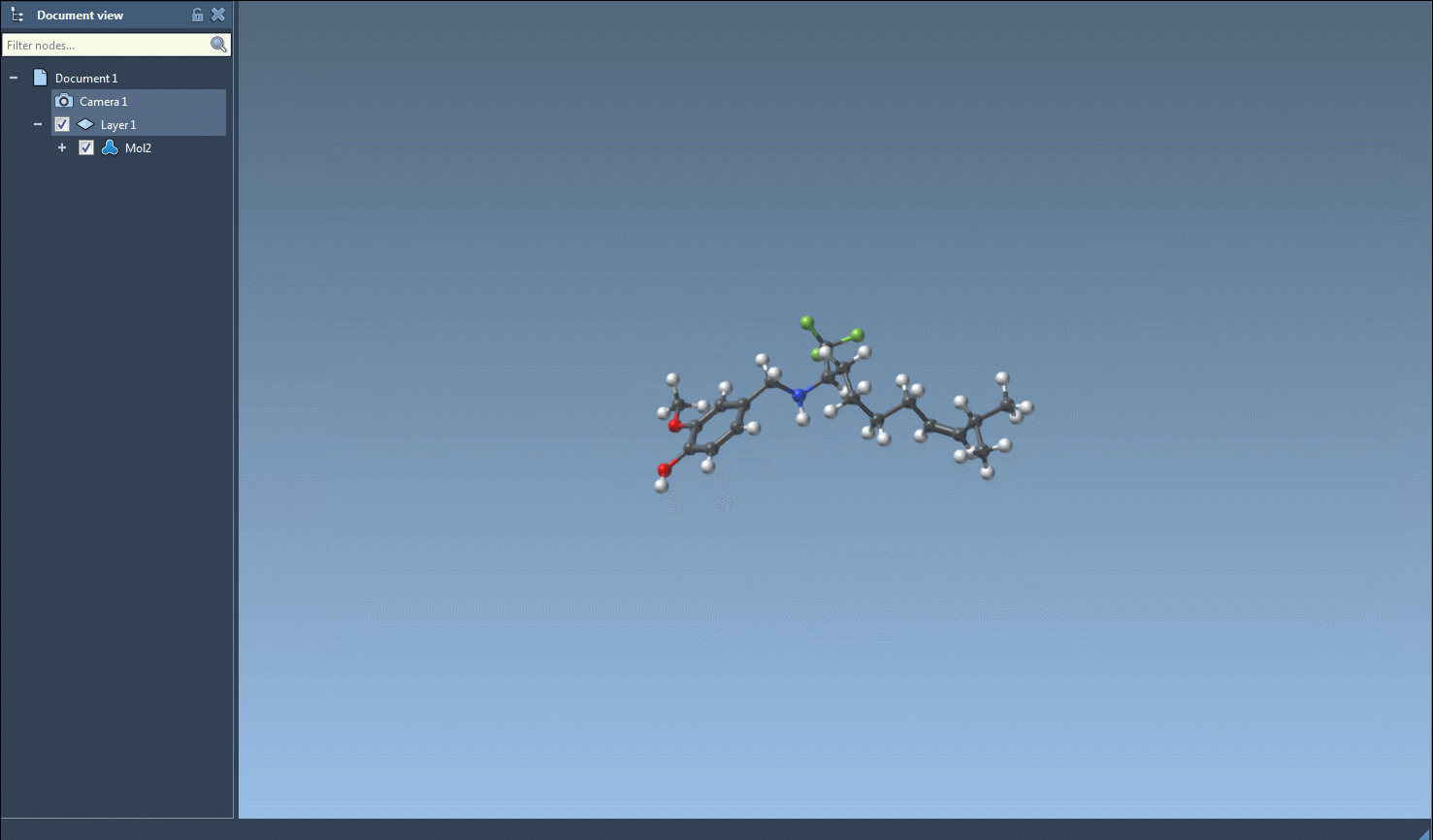 Note that by default, in RDKit, information about stereochemistry is not used in substructure searches but this can be changed by enabling the chirality.
Note that by default, in RDKit, information about stereochemistry is not used in substructure searches but this can be changed by enabling the chirality.
Replace fragments#
The Replace fragments tab allows for quick and easy generation of thousands of different molecules by replacing a specified fragment of the initial molecule with various fragments. This feature provides isosteric replacement and can be useful for quickly generating a library of slightly different molecules to test their properties.
Note that each table is presented in the same way as the SMILES table in the Manage SMILES tab with the same buttons. One difference is the saving of the table as a grid or as a file which is present as buttons at the bottom right of each table.
Let's try to generate a library of fragments for a molecule:
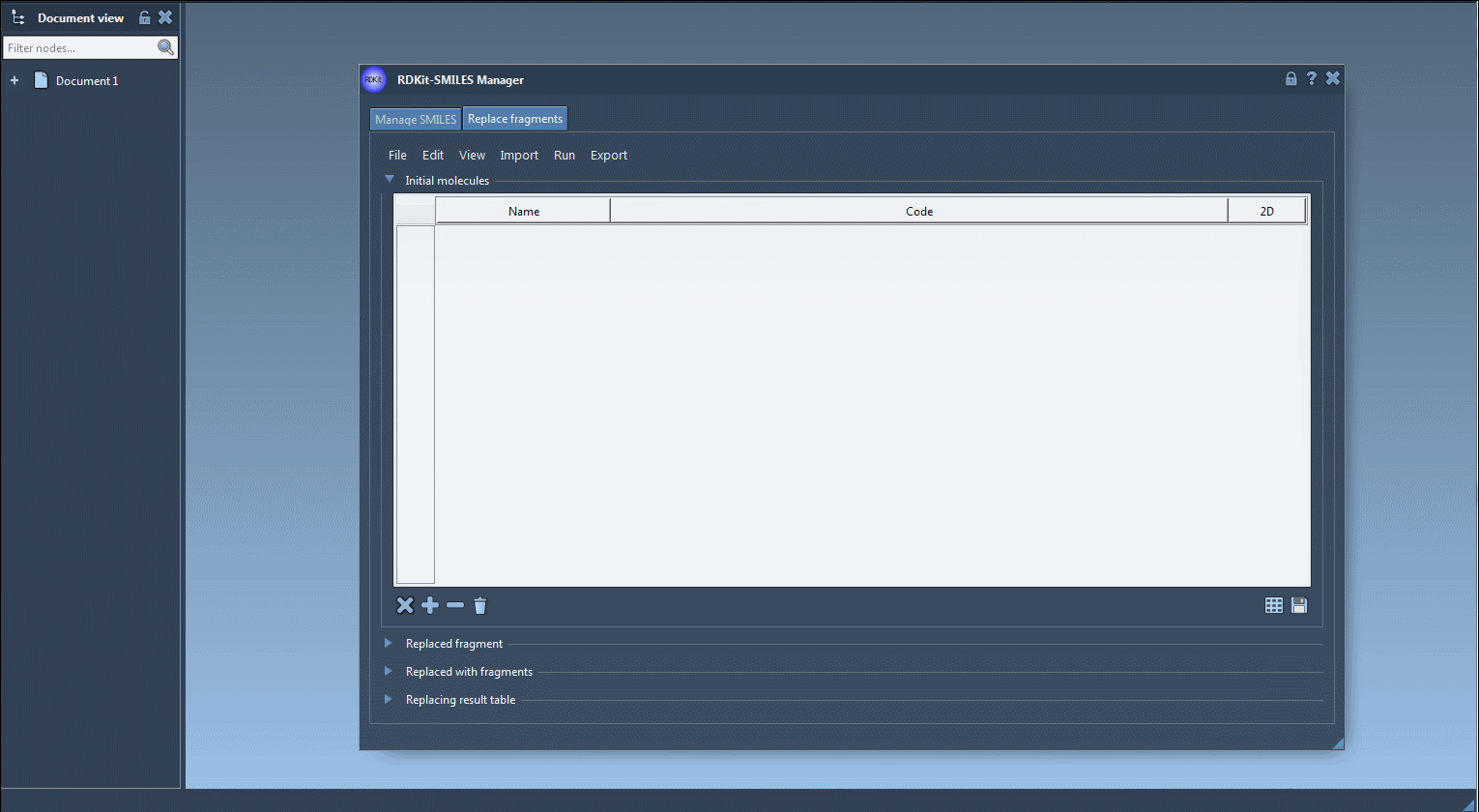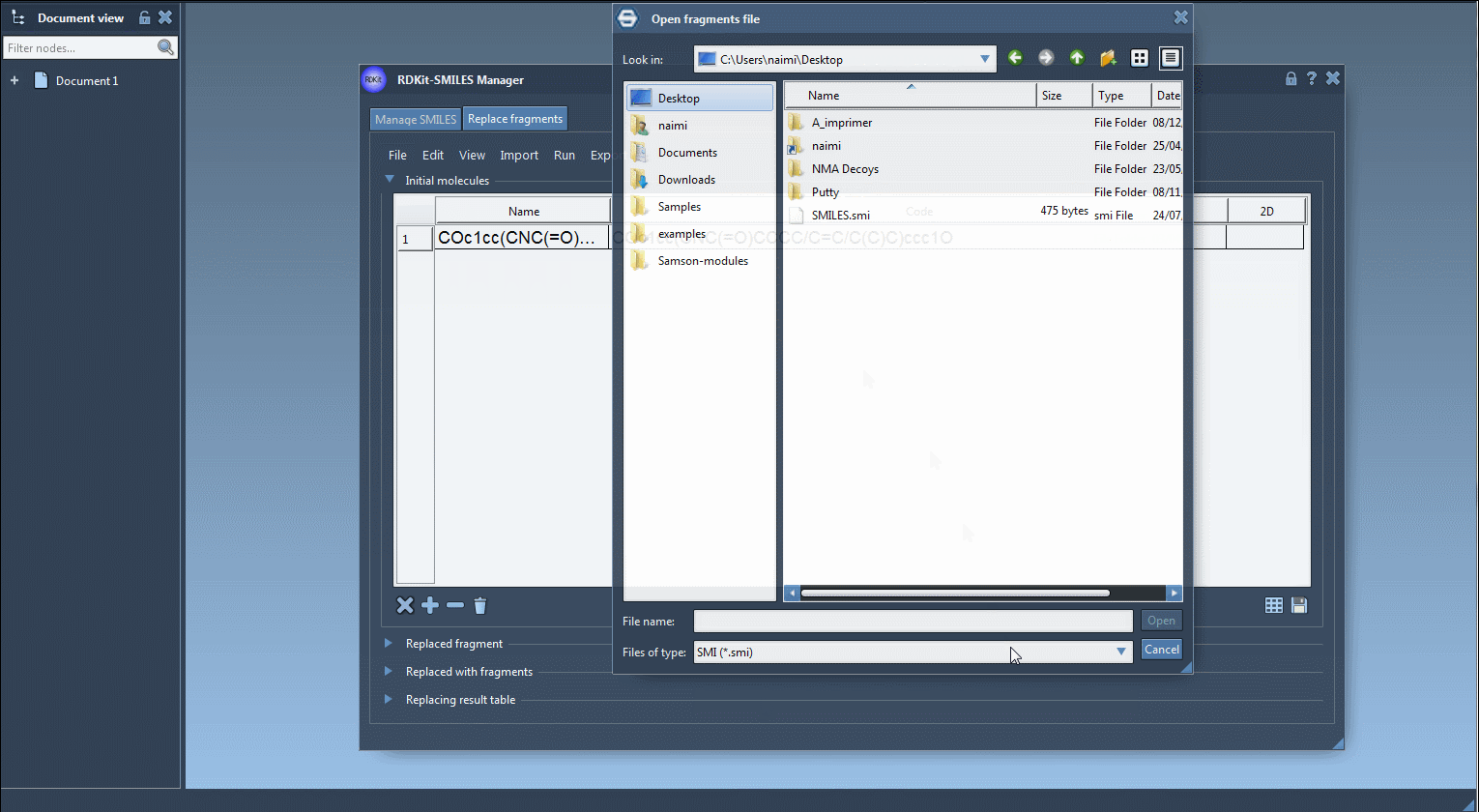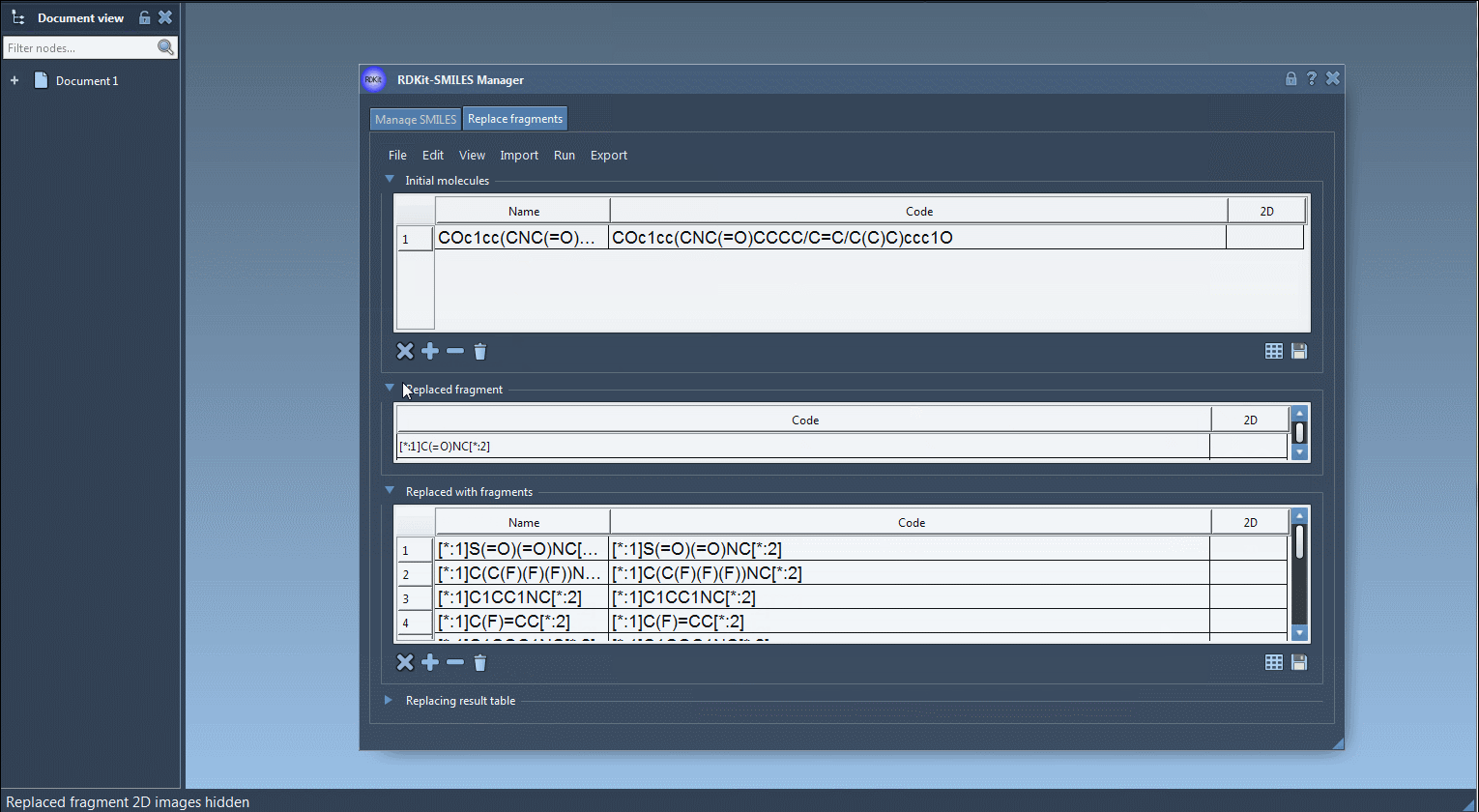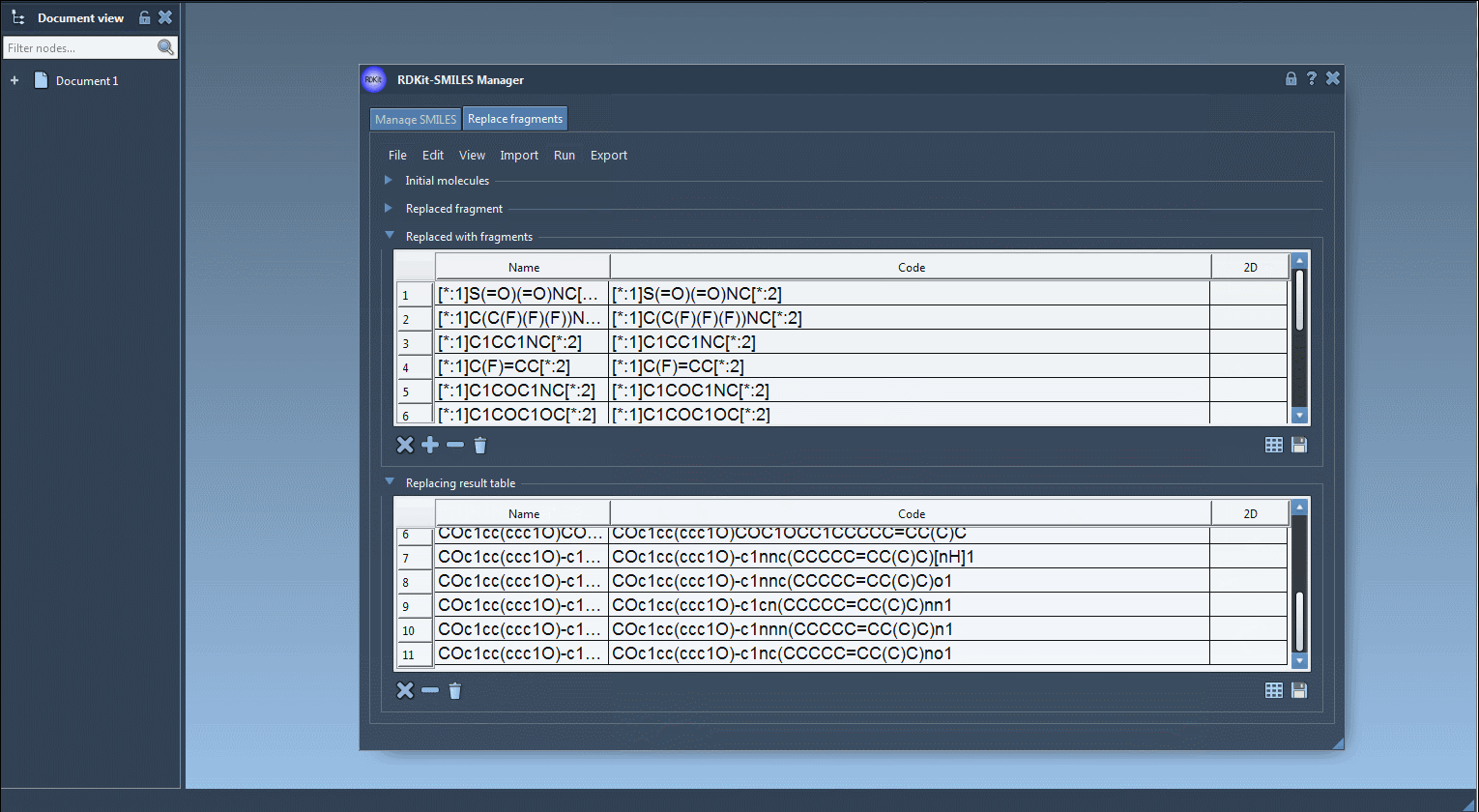
- First, we need to specify one or several initial molecules in which we would like to replace a fragment. You can import initial molecules from a file by clicking Open initial molecules in the File drop-down menu, add them manually, import from a SAMSON Document by clicking SAMSON Selection as initial molecule in the Import drop-down menu. In this tutorial, we will use the following initial molecule (load it from a file or add it manually):
COc1cc(CNC(=O)CCCC/C=C/C(C)C)ccc1O - Enter the fragment that you want to be replaced in the initial molecules either by entering it manually or by importing it from the active SAMSON Document by going to the SAMSON Selection as replaced fragment action in the Import drop-down menu. In this tutorial, the target fragment to replace is:
[*:1]C(=O)NC[*:2] -
Provide one or more "Replaced with fragments" (fragments with which you want to make a replacement) by importing from a file (click Open fragments to replace with... in the File drop-down menu) or by adding them manually. In this tutorial, we will be using the following fragments:
[*:1]S(=O)(=O)NC[*:2] [*:1]C(C(F)(F)(F))NC[*:2] [*:1]C1CC1NC[*:2] [*:1]C(F)=CC[*:2] [*:1]C1COC1NC[*:2] [*:1]C1COC1OC[*:2] [*:1]C1=NN=C([*:2])N1 [*:1]C1=NN=C([*:2])O1 [*:1]N1N=NC([*:2])=C1 [*:1]N1N=NC([*:2])=N1 [*:1]C1=NOC([*:2])=N1 -
In the Run drop-down menu, click on the Replace fragments.
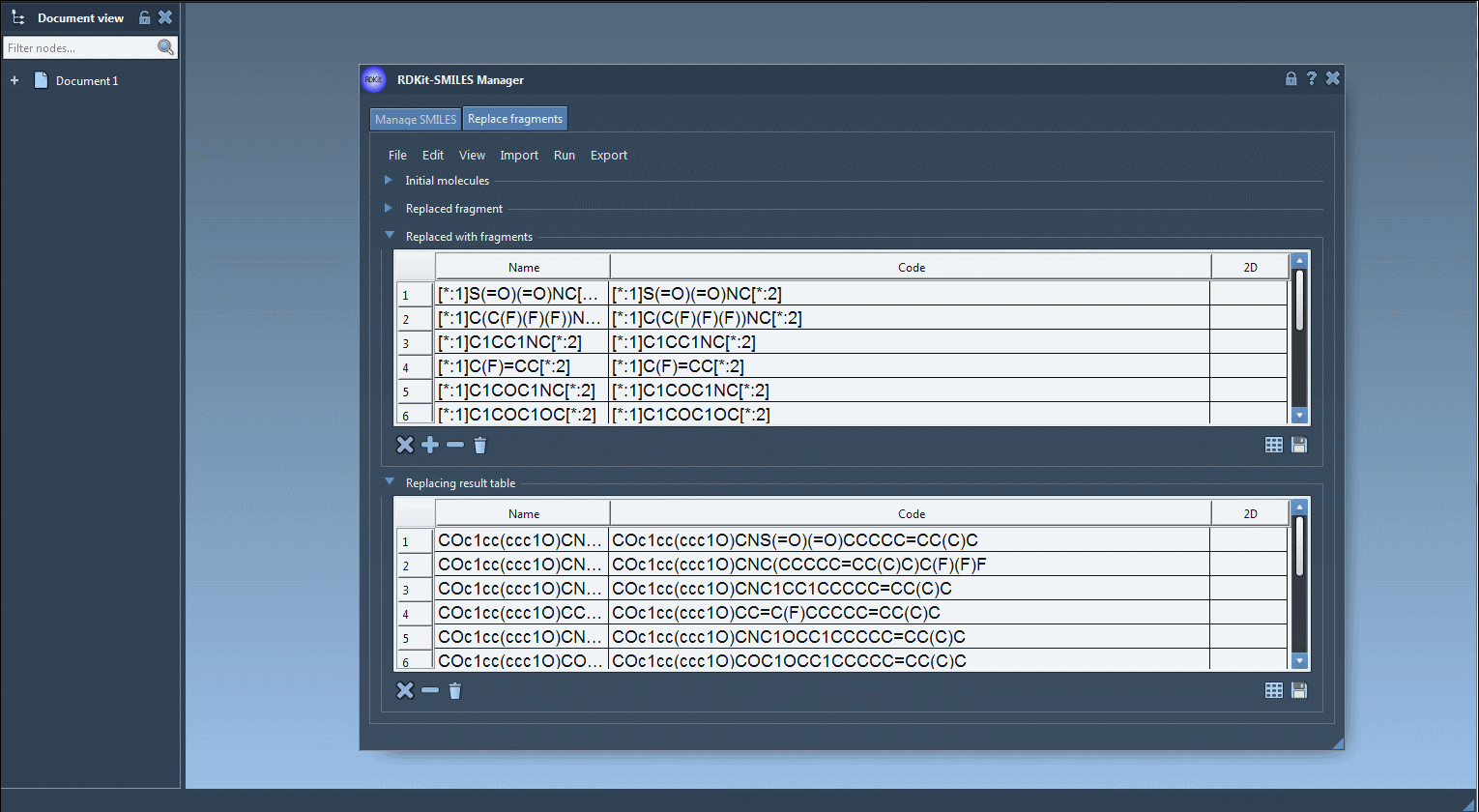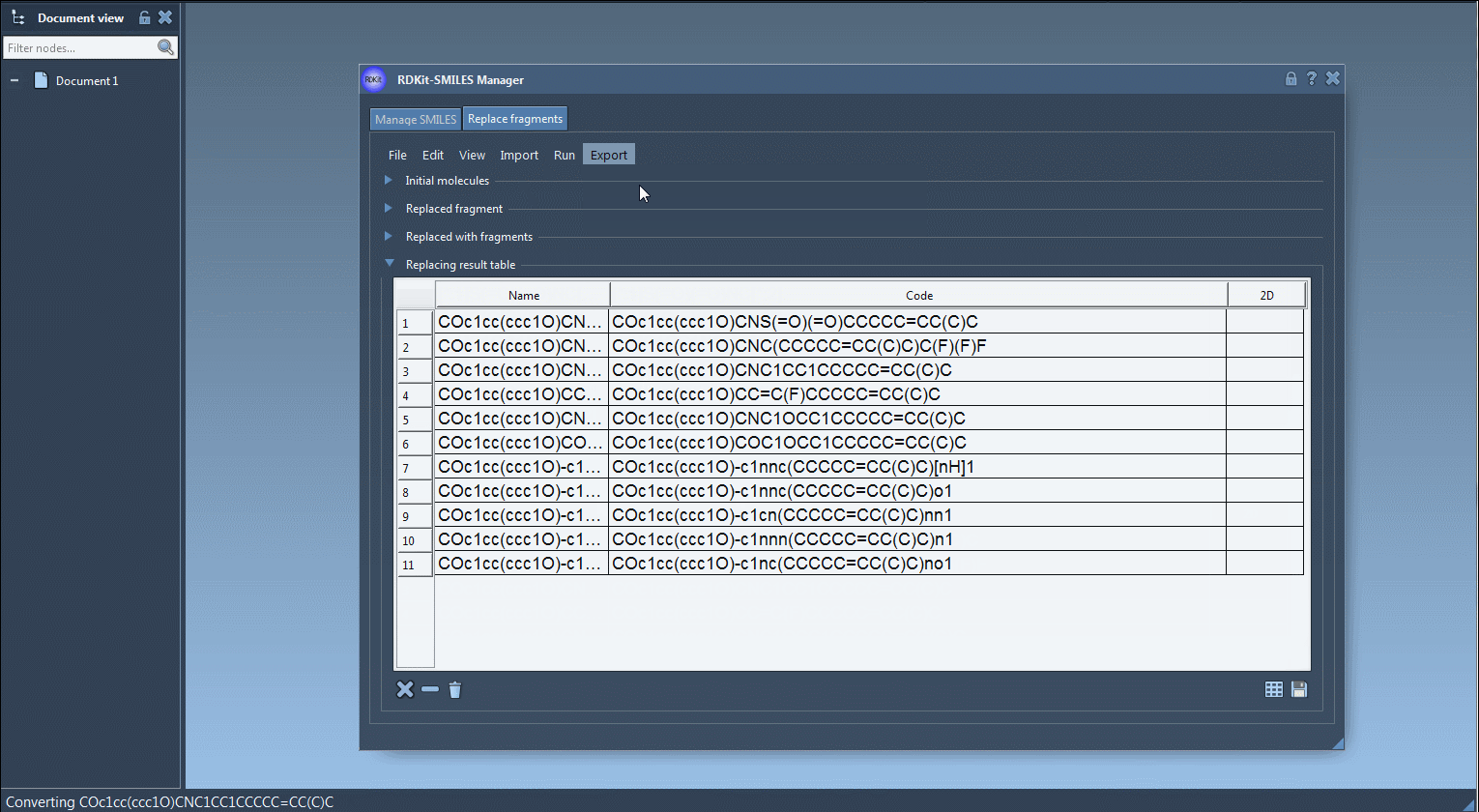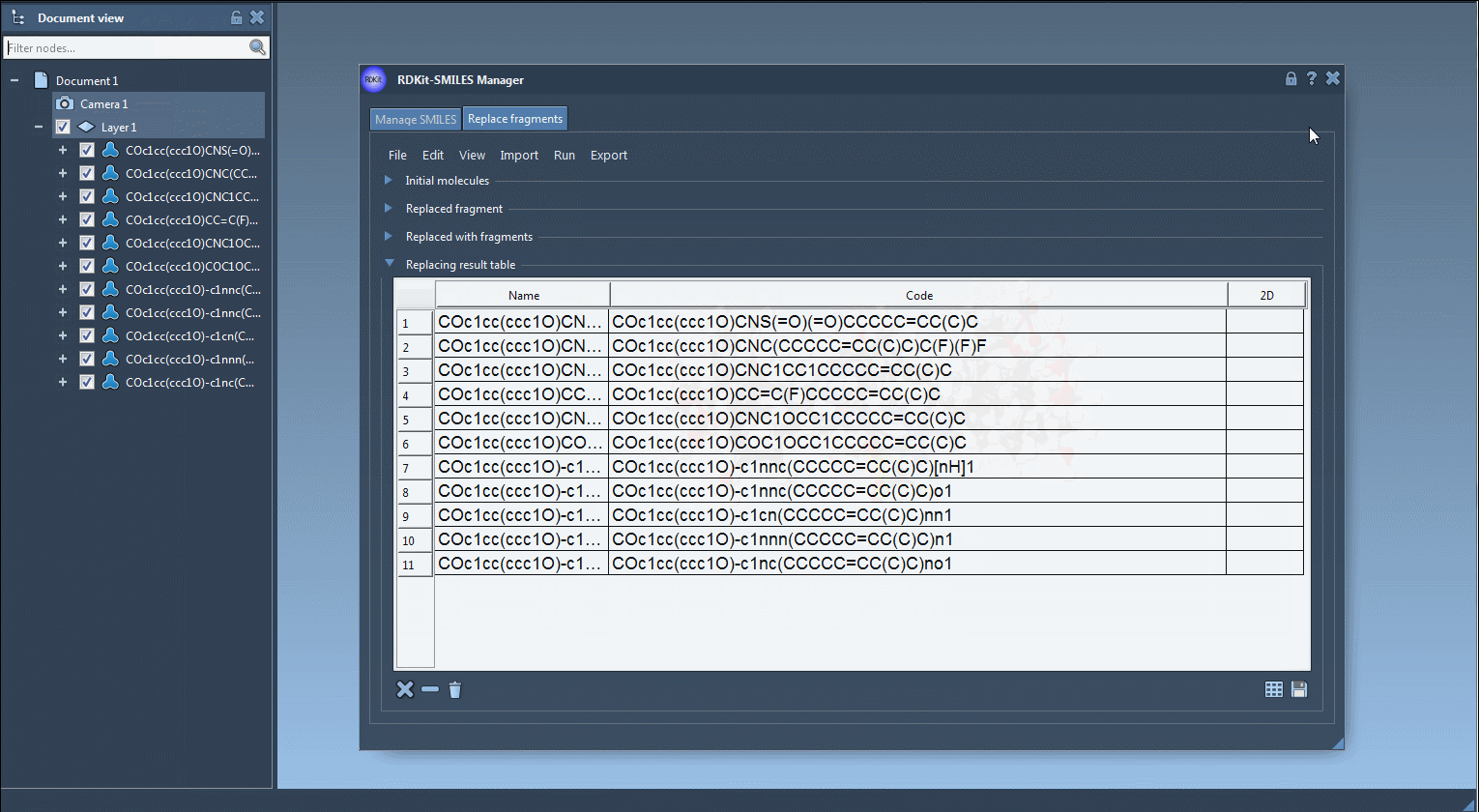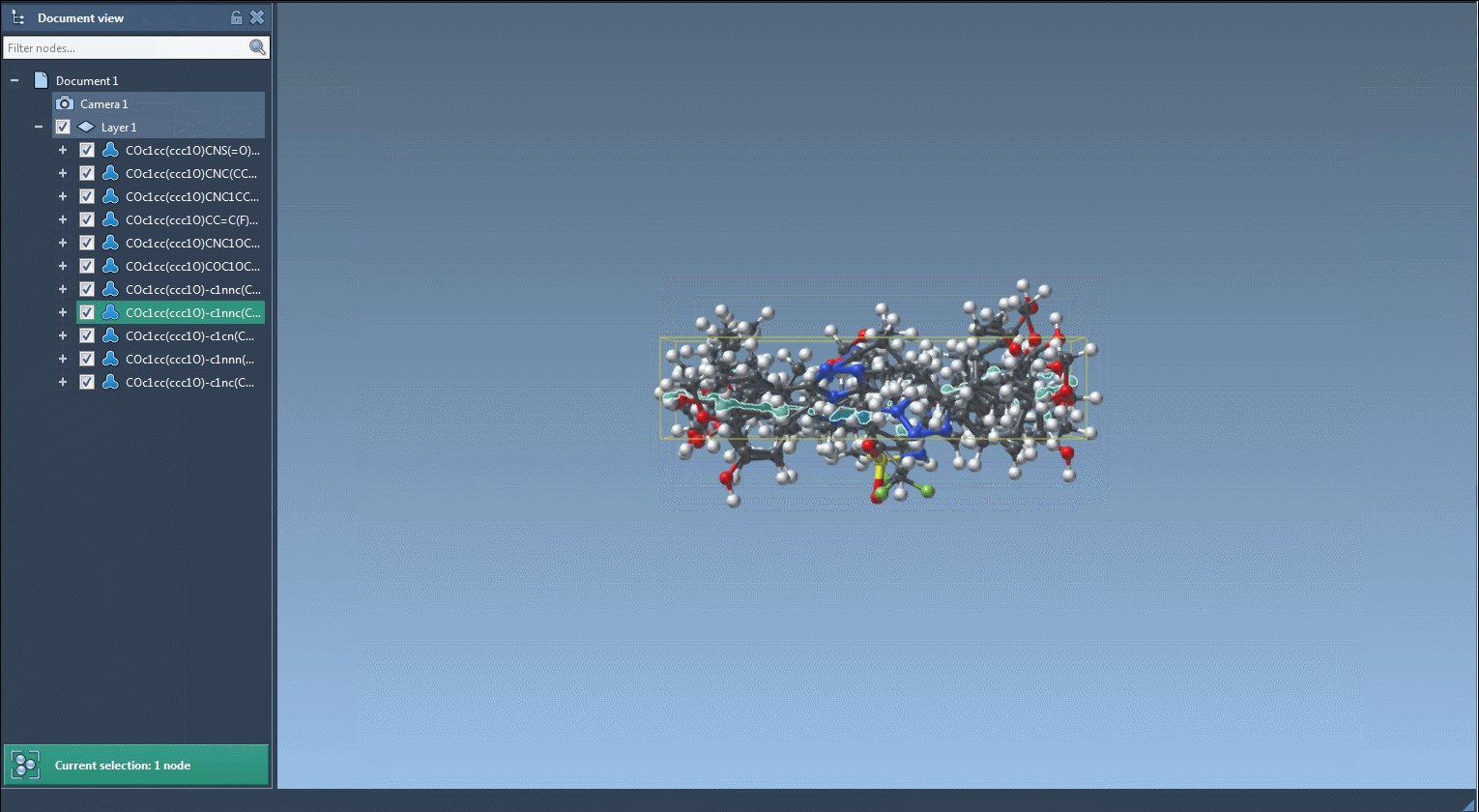
- Results of the replacements are stored in the Results table. You can view them in the active document - check out the Export drop-down menu.
Note that in RDKit the fragments must have the following syntax: [*:1]C(=O)NC[*:2] with [*:1] and [*:2] at the two extremities of the pattern.
The resulting molecules can then be converted into 3D structures, modified, or saved in a file or a grid image.
That's all for this tutorial about the SMILES Manager module of SAMSON. You can check out the Positional Analogue Scanning using the SMILES Manager Extension tutorial.
Related tutorials#
- Generate analogue series with positional analogue scanning for a focused analogue-generation workflow.
- Dock ligands and libraries with AutoDock Vina Extended for docking generated or imported molecules.
- Strain Explorer for analyzing generated ligand conformations.
Next step#
Use the SMILES Manager results to generate 3D structures, build analogue series, or prepare molecules for docking and analysis.
Need Help?#
Have questions or feedback? Feel free to reach out via the Forum, via e-mail, via the Feedback button in SAMSON, or by directly discussing with us.